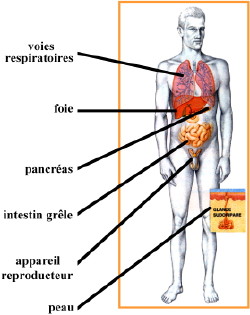
 La mucoviscidose est la maladie génétique récessive autosomale (portée par un chromosome non sexuel) la plus répandue dans les populations européennes et nord-américaines. Environ un enfant sur 3500 naît atteint de cette maladie avec une espérance de vie de 35 à 50 ans selon les pays. Les organes ciblés par la mucoviscidose sont nombreux (figure ci-contre).
La mucoviscidose est la maladie génétique récessive autosomale (portée par un chromosome non sexuel) la plus répandue dans les populations européennes et nord-américaines. Environ un enfant sur 3500 naît atteint de cette maladie avec une espérance de vie de 35 à 50 ans selon les pays. Les organes ciblés par la mucoviscidose sont nombreux (figure ci-contre).
Les mutations identifiées sur le gène CF (Cystic Fibrosis, terminologie anglo-saxonne de la mucoviscidose) sont associées à la mucoviscidose. Ce gène code une protéine transmembranaire appelée CFTR (Cystic Fibrosis Transmembrane Regulator) qui assure à la fois la sécrétion passive des ions chlorures (Figure) et la régulation d’autres transporteurs. Le dysfonctionnement de CFTR provoque l’épaississement du mucus et son accumulation dans les voies respiratoires et digestives offrant ainsi un terrain propice aux infections opportunistes principalement dans les poumons. Soixante dix pour cent des mutations identifiées sur le gène CF correspondent à la délétion d’un acide aminé au niveau de la protéine CFTR (une phénylalanine en position 508 d'où le nom delF508). Cette mutation est responsable du blocage et de la dégradation rapide au niveau du réticulum endoplasmique d’une protéine CFTR mal repliée mais fonctionnelle. Ainsi, corriger ce défaut d’adressage dans les cellules malades pour rétablir la sécrétion des ions chlorures est devenu une cible thérapeutique privilégiée. Cependant, l’efficacité de ces thérapies se trouve diminuée par la rapidité de dégradation du CFTR-delF508 dont les mécanismes restent très débattus.
Tout récemment, des chercheurs de notre laboratoireont montré que le nucléotide GTP joue un rôle très important dans la dégradation du CFTR-delF508 et d’autres protéines mal repliées. Dans leur travail, ces chercheurs ont caractérisé biochimiquement cette nouvelle voie de dégradation ainsi que les protéines potentiellement impliquées.
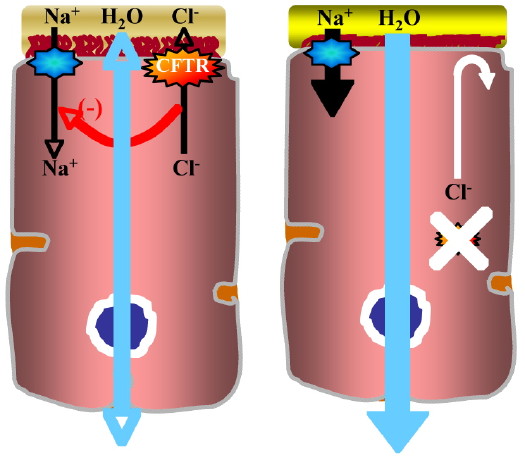
Figure : Transport hydro-électrolytique dans une cellule épithéliale pulmonaire normale et mucoviscidosique.
Les cellules épithéliales pulmonaires ciliées normales (gauche) sont tapissées par un mucus dans la viscosité est maintenue garce à un équilibre entre la réabsorption du sodium (Na+), la sécrétion des ions chlorures (Cl-) assurée par la protéine CFTR et le transport trans-épithélial de l’eau. Dans une cellule malade (droite), l’absence de sécrétion des ions chlorures provoque une augmentation de la réabsorption du Na+ et de l’eau engendrant ainsi l’épaississement du mucus qui va détériorer le tapis cilié et offrir un terrain propice aux infections opportunistes.
C’est la première fois que des chercheurs démontrent la contribution du GTP dans la voie de dégradation associée au réticulum endoplasmique. Déterminer la nature moléculaire et les mécanismes protéolytiques de cette voie de dégradation permettra à long terme de mieux cibler la pharmacothérapie afin de rétablir la sécrétion normale des ions chlorures chez les malades atteints de mucoviscidose.
Le réticulum endoplasmique est un compartiment intracellulaire lieu de synthèse et de repliement des protéines transmembranaires et de sécrétions.