Large molecular assemblies - Membrane protein simulations - Coarse graining
Published on 25 January 2019
Body text 1
Simulations of proteins in an explicit aqueous medium for a period of one hundred nanoseconds are currently possible. However, the dynamic and the most relevant interactions within cells (protein-protein docking, rearrangement following the binding of a ligand or after a biochemical reaction, folding) occur on time scales exceeding the millisecond, and involve large macromolecular aggregates. The simulation of such systems is not currently feasible with all-atom models. In addition, in some cases, simulations at the atomic level may not be the most appropriate for an experimental validation because a growing number of biomolecular aggregates are studied using techniques from low to medium resolution (such as cryo-electron microscopy and X-ray small angle scattering). Thus, the idea of using simplified descriptions through "integration" of a large number of degrees of freedom in a “coarse grain” model seems natural. However, the parameterization of these “grains” can be difficult. We chose to use known and validated models (MARTINI) in our simulations, an example of which is shown in the following figure.



A. Model of calcium ATPase inserted in a phospholipid bilayer all atom model.
B. “Coarse” representation of the same system. Each amino acid is simplified into 2 or 3 spheres, the polar heads of the phospholipids in 5 and the hydrophobic in 6 spheres. Spheres interact with each other via a Martini type potential modified for proteins.
C. Coarse grain model of the ATPase alone in Sansom.
 Signal transduction mechanism leading to the opening of an ionic channel
Signal transduction mechanism leading to the opening of an ionic channel
Karthik Arumugam), Nicolas Sapay, Serge Crouzy (collaboration with Michel Vivaudou and C Moreau, IBS Grenoble
We work with M. Vivaudou's team on the ANR ICCR (Ionic Channel Coupled Receptors) project. An ICCR is a membrane receptor (GPCR: G protein receptor) coupled to an ion channel to form an artificial channel whose opening is controlled by the receptor ligand. The first ICCR was built in the IBS team by engineering fusion proteins between the potassium channel Kir6.2 and the human muscarinic receptor: HM2 [1]. The conformational changes resulting from the binding of acetylcholine (ACh) to HM2 are transmitted to Kir6.2 and the resulting ionic current (pA) is detected with electrophysiological techniques.
Using a modeling approach, as explained for instance in [2], we aim at a rational approach to the construction of new functional fusion proteins.
Nicolas Sapay's work during his Post-doc in our team was to predict the structures of the partners (when unknown), position the GPCR relative to the channel, build the functional tetramer, then find a possible mechanism of signal transduction between GPCRs and channel leading to its opening (or closing).
The following figure shows a model structure of HM2 coupled to Kir6.2.
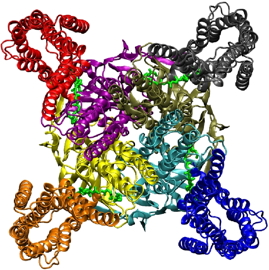
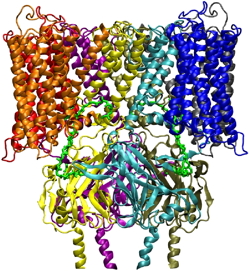 Model structure of the human HM2 G protein-coupled receptor linked to the Kir6.2 potassium channel. Each monomer of the tetramer is shown in a different color:
Model structure of the human HM2 G protein-coupled receptor linked to the Kir6.2 potassium channel. Each monomer of the tetramer is shown in a different color:
A: view from below, cytoplasmic side.
B: front view.
Interactions of Arabidopsis thalliana HMA6 and HMA8 copper ATPases with plastocyanin
Collaboration with Patrice Catty and Daphné Seigneurin-Berny
Copper is an essential metal ion for the plant with key roles in many cellular processes. In Arabidopsis, approximately 50% of the cellular copper is located in the chloroplast, mostly associated with copper superoxide dismutase (Cu / Zn SOD) of the stroma, and with plastocyanin (PC) of thylakoids. Since plastocyanin is essential for photosynthesis, addressing of copper to chloroplasts is therefore prioritized by the cell. In Arabidopsis, three P-type ATPases, HMA1, HMA6 and HMA8, transport copper into the chloroplast. HMA6 located in the envelope is the main channel of copper import. It allows to power the SOD then the PC via HMA8. Could there be a direct interaction between HMA8 and plastocyanin? This is the question we have attempted to answer through modeling. We performed a modeling of HMA6 and HMA8 by homology with Legionella pneumophila copper ATPase (PDB 4BBJ). Then, a docking between HMA8 and the plastocyanine was carried out with the HADDOCK software. The result is shown in the following figure: there could be a possibility of transferring the ATPase metal (via the CPC path, M857 D282 ...) to the copper bonding motif (H37, H84, H87) of the PC [3, 4].
01 - Moreau C, Dupuis JP, Revilloud J, Arumugam K and Vivaudou M
Coupling ion channels to receptors for biomolecule sensing.
Nature Nanotechnology, 2008, 3(10): 620-625
02 - Crouzy S
Dynamique moléculaire et canaux ioniques.
Journal de Physique IV, 2005, 130: 179-191
03 - Sautron E, Mayerhofer H, Giustini C, Pro D, Crouzy S, Ravaud S, Pebay-Peyroula E, Rolland N, Catty P and Seigneurin-Berny D
HMA6 and HMA8 are two chloroplast Cu+-ATPases with different enzymatic properties.
Bioscience Report, 2015, 35(3): e00201
04 - Sautron E, Giustini C, Dang T, Moyet L, Salvi D, Crouzy S, Rolland N, Catty P and Seigneurin-Berny D
Identification of two conserved residues involved in copper release from chloroplast PIB-1-ATPases.
Journal of Biological Chemistry, 2016, 291(38): 20136–20148
Top page