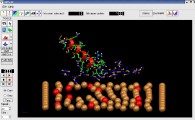
 Ce projet a été écrit en collaboration avec Stéphane Redon de l'INRIA et Michel Vivaudou. Le but était de développer un outil de visualisation interactive et de nouveaux algorithmes pour l'étude dynamique et la manipulation interactive de macromolécules biologiques à partir du travail de Stéphane Redon sur les objets articulés, de champs de forces simplifiés pour les interactions atomiques et de leur visualiseur MPV3D (Figure ci-contre).
Ce projet a été écrit en collaboration avec Stéphane Redon de l'INRIA et Michel Vivaudou. Le but était de développer un outil de visualisation interactive et de nouveaux algorithmes pour l'étude dynamique et la manipulation interactive de macromolécules biologiques à partir du travail de Stéphane Redon sur les objets articulés, de champs de forces simplifiés pour les interactions atomiques et de leur visualiseur MPV3D (Figure ci-contre).
En raison de la complexité importante des structures et interactions qu'ils étudient, les biologistes utilisent fréquemment des représentations simplifiées de la géométrie et de la dynamique des molécules impliquées. Fréquemment, ces méthodes de simplification consistent à utiliser des représentations en coordonnées réduites (e.g. modélisation dans l'espace des angles), et à remplacer des sous-ensembles d'atomes par des structures idéalisées.
Parce qu'elles contiennent moins de degrés de liberté, ces structures simplifiées permettent d'accélérer le calcul de la dynamique de la molécule, et facilitent l'étude des interactions moléculaires. Elles permettent également d'obtenir plus efficacement des structures d'énergie minimale qui tiennent compte des contraintes expérimentales. Cependant, le problème fondamental des méthodes de simplification actuelles est qu'elles sont incapables de déterminer automatiquement le niveau de détail le mieux à même de représenter une interaction moléculaire donnée. Ainsi, le biologiste doit avoir une idée a priori de l'interaction qu'il souhaite modéliser avant de choisir la représentation la mieux adaptée. Autrement dit, il n'existe pas, à l'heure actuelle, de méthode permettant de déterminer automatiquement quelles parties de la molécule doivent être simulées de façon précise, et quelles parties peuvent être simplifiées sans nuire à l'étude de l'interaction moléculaire.
 Le simulateur de dynamique moléculaire se devait donc d'être adaptatif, interactif et multi échelle.
Le simulateur de dynamique moléculaire se devait donc d'être adaptatif, interactif et multi échelle.
• Adaptatif, car le simulateur détermine automatiquement les zones de la molécule les plus pertinentes qui doivent être simulées (cela permet de diminuer de façon considérable les temps de calcul, tout en conservant des résultats fiables) (Figure ci-dessus).
• Multi échelle, puisque le simulateur utilise différents niveaux de détail à l'intérieur d'une même molécule.
 • Interactif, car il est couplé à un bras à retour de force permettant une manipulation efficace et intuitive des molécules (Figure ci-contre).
• Interactif, car il est couplé à un bras à retour de force permettant une manipulation efficace et intuitive des molécules (Figure ci-contre).
La première application visée était l'étude des transporteurs ABC, transporteurs de métaux et autres canaux ioniques, cibles majeures dans le domaine pharmaceutique, en raison de leur implication dans de nombreux processus physiologiques et pathologiques. Il sera également intéressant de pouvoir proposer des modèles simplifiés permettant, par exemple, de calculer efficacement l'action de forces extérieures simulant l'ouverture d'un canal ionique, et d'évaluer l'impact de ces forces sur la dynamique du modèle. Par extension, d'autres applications pourront être envisagées comme le « docking » (amarrage moléculaire) ou le repliement des protéines.