Modelling, structure prediction reactivity and dynamics of metalloproteins
Published on 25 January 2019
Body text 1
 Introduction
Introduction
Modeling, the use of a simplified model to explain a phenomenon, has three main applications for us in biology and chemistry:
• Structures refinement.
¬ In NMR from interatomic distances (2-5 Å) and dihedral angles.
¬ In X-ray crystallography from the diffraction patterns.
• Structure prediction
¬ Prediction of secondary or tertiary structure of proteins from their sequence.
Homology models.
¬ Extraction of time frames of the evolution of a system simulated using classical MM as a basis for a study of type QM / MM.
• Simulation: a numerical in silico experiment complementary to conventional in vitro or in vivo experiments to:
¬ calculate experimental observables and try to deduce the mechanisms that determine them.
¬ obtain measures beyond the experimental possibilities in perfectly controlled conditions of purity, pressure, temperature ..
¬ test our understanding of phenomena at the atomic scale.
¬ visualize the consequences of thermal agitation of macromolecular structures (access of substrates to enzymatic sites, conformational transitions, the influence of water, coupling between movements.)
Our research covers the three previous topics but the simulation of macromolecules in order to understand some aspects of their function from their structure is predominant.
In a conventional MM approach, any molecule being a combination of various atoms, the cohesion of this set results in repulsive and attractive interactions between atoms and will be modeled by a purely mechanical system of forces, without taking into account the microscopic and electrostatic nature (quantum) of the interactions between electrons and nuclei.
A macromolecule is thus modeled by a network of point masses charged in their center, connected by springs and interacting via empirical potentials.
Follow this link for a brief introduction to the molecular mechanics (MM) and dynamics (MD)
We present in the following a few examples of our work in progress in three main areas:
In quantum mechanics-based methods (including density functional theory), the system of interest such as an enzymatic active site is modelled at the electronic level.
Finally, in a so called QM/MM approach, a large part of the macromolecule is treated in MM and even partially frozen, while the site of interest is treated in QM.
Metallo-chaprones / ATPases interactions and metal transfer
Serge Crouzy, Yohann Moreau
Former PhD students: David Poger, Karthik Arumugam
We are interested in the specificity and affinity of the interaction of various metal ions (Cu+, Hg2+, Cd2+, Pb2+) for Cu(I) metallochaperones (Atx1 in yeast and Hah1 in humans ) and their associated (Ccc2 and Menkes, respectively) ATPases. The study of the dynamics of the metallochaperone Hah1 and selectivity of the binding of Cu+ and Hg2+ to the protein was carried out by D. Poger during his thesis in the laboratory [1, 2]. We demonstrated that the consensus sequence MxCxxC present in Hah1 and model peptides among many metal transporters binds Hg(II), Cu(I), Pb(II), Cd(II), and Zn(II) but not Cu(II) [3]. An interesting feature of the P1-type ATPase is the presence at their N-ter of one to six metal binding domain(s) (MBDS). In the case of the Menkes ATPase, there are 6 MBDS, all able to bind a Cu+ ion. The structure of each MBD is known. Using MD simulations, we studied the dynamics of each MBD in the presence or absence of metal in order to understand how the metal is transferred from the metallochaperone in charge of the metal during its entry into the cell to the MBD. These studies have used our work in the field of parameterization of molecular mechanics force fields for metal ions [4]. The following figures (See the following A and B figures) show possible interactions between the soluble Hah1 metallochaperone and the first metal-binding domain (Mnk1) of its membrane partner Menkes ATPase.
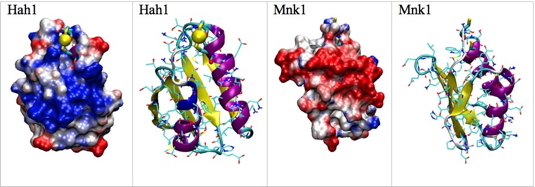
A - Electrostatic potential maps and secondary structure, in the same orientation, for Hah1 and the binding domain 1 (Mnk1) of the Menkes ATPase. The two proteins have been rotated by 90 degrees to present their interacting surfaces. Hah1 contains a copper ion (yellow sphere) which is supposed to be transfered to Mnk1 in its Apo state.
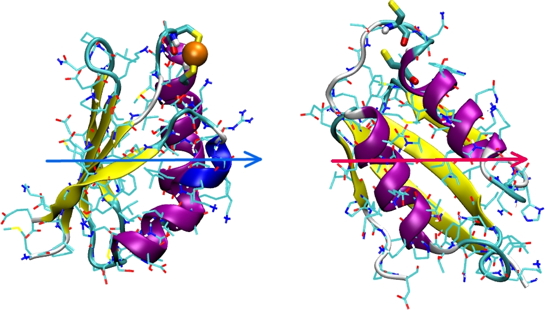
B - Representations of the metallochaperone Hah1 and the first domain of the Menkes ATPase (Mnk1) as they should interact (reconciled) in the case of a dominant electrostatic interaction. Electric dipoles of the two proteins are represented
The good agreement of our MD results on MBDs with experimental data [5] has shown that mechanical models are able to account for and explain some properties of binding and transport of metals in metalloproteins and ATPases, which are well known drug targets.
Design and functionalization of metalloproteins
Yoann Moreau, (Collaboration with Vincent Forge, Nicolas Durrafourg, team LCBM/AFFOND)
Former students: Justine Lévêque and Magda Benisz
The increasing interest for nanotechnologies can lead to imagine the possibility of modifying natural structures such as proteins to give them selected properties. It is thus possible, by the mean of directed mutagenesis, to modify a part of the structure so as to create an active site with controlled properties. In collaboration with team AFFOND at LCBM, we have studied the possibility to rebuild an active site in given proteins. Theoretical studies, based on quantum models have particularly shown that it was possible, by simply mutating chosen residues, to “rebuild” the active site of plastocyanin with most of its structural and electronic properties. This project was initiated by the work of J. Levêque and M. Benisz.
Modelling the inhibition of the regulator FUR by peptide aptamers
Thesis of Serge Nader
Former student: Cheickna Cissé (Collaboration with I. Michaud Soret and J. Pérard in the BioMet team)
The bacterial ferric uptake regulator (FUR) is an essential iron responsive protein that controls iron homeostasis and virulence gene transcription. Its inactivation can result in decreased pathogenic virulence. In I. Michaud-Soret's team, specific FUR inhibitors were isolated by the technology of peptide aptamers. Four peptide aptamers (F1 to F4) comprising an E. coli thioredoxin A scaffold and a 13 amino acid variable loop were identified as interacting with E. coli FUR, inhibiting its transcriptional function and decreasing its virulence. The goal of our research is to understand this inhibition mechanism and deduce other inhibitory molecules for therapeutic applications. Modeling studies and biochemical interaction are taken to achieve this end.
In the MCT team, the study of the interactions between FUR and its inhibitors is done by molecular docking with AUTODOCK. This study already allowed us to find regions of the inhibitors and amino acids of the protein in possible interaction. The homology models of the FUR protein were constructed with MODELLER in the presence of iron II metal ions. Interactions found by docking were validated by experimental binding assays of FUR to DNA.
A good peptide inhibitor of FUR (denoted pF1 from the F1 aptamer) was early proposed, it has excellent theoretical affinity for the activated form of FUR.
In this configuration, the inhibitor interacts with the amino acids of a protein pocket, inhibiting the binding of FUR to DNA and acts on the two domains of the protein (see following figure). This result is consistent with previous work done with the two-hybrid technique on peptide aptamers indicating that this inhibitor requires the two domains of the protein to interact.
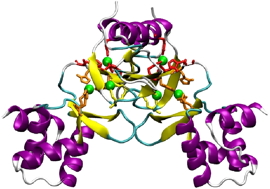
Left: Model of E coli FUR by homology with FUR from vibrio cholerae in the presence of three metal atoms per monomer (green spheres) and
Right: 3D representation of the pF1 inhibitor docked on the model of the FUR protein with Autodock. (Representation with VMD).
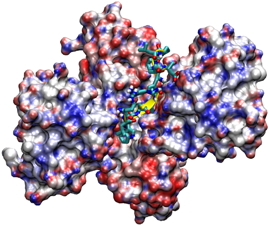
More recently, the F2 variable loop has been studied as the linear peptide pF2 (RQCNICGASLYSY). We investigated the inhibitory properties of pF2 and characterized its interaction with the FUR dimerization and DNA binding domains. pF2(1−11) and pF2(1−10) inhibit FUR more strongly than pF2 itself, while the shortest active peptide is pF2(2−10), eliminating R1 results in a 9-fold IC50 increase. The E. coli FUR residues S126 and Y128 are located in the binding pocket and are essential for pF2 interaction. Using docking experiments we showed that two pF2 peptides can simultaneously bind separate sites on the E. coli FUR dimer, one per each subunit. The bound peptides sterically hinder the accessibility of DNA to its binding domain on FUR, effectively inhibiting the protein (see following figure). Our collaborators in the BioMet team used isothermal calorimetry to determine that pF2 has a submicromolar dissociation constant, while Autodock returned a very high binding energy with ΔG=-23 kcal/mol for pF2(1-11).
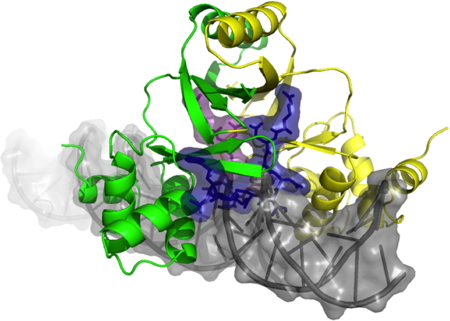
Overlap of the model of EcFUR-ΔCter15 dimer complexed to two pF2(1-11) peptides and the EcFUR-ΔCter15 dimer interacting with DNA showing the steric hindrance.
01 - Poger D, Fuchs JF, Nedev H, Ferrand M and Crouzy S
Molecular dynamics study of the metallochaperone Hah1 in its apo and Cu(I)-loaded states: Role of the conserved residue M10.
FEBS Letters, 2005, 579(24): 5287-5292
02 - Poger D, Fillaux C, Miras R, Crouzy S, Delangle P, Mintz E, Den Auwer C and Ferrand M
Interplay between glutathione, Atx1 and copper: X-ray absorption spectroscopy determination of Cu(I) environment in an Atx1 dimer.
Journal of Biological Inorganic Chemistry, 2008, 13(8): 1239-1248
03 - Fuchs JF, Nedev H, Poger D, Ferrand M, Brenner V, Dognon JP and Crouzy S
New model potentials for sulfur-copper(I) and sulfur-mercury(II) interactions in proteins: From ab initio to molecular dynamics.
Journal of Computational Chemistry, 2006, 27(7): 837-856
04 - Rousselot-Pailley P, Seneque O, Lebrun C, Crouzy S, Boturyn D, Dumy P, Ferrand M and Delangle P
Model peptides based onthe binding loop of the copper metallochaperone Atx1: Selectivity of the consensus sequence MxCxxC for metal ions Hg(II), Cu(I), Cd(II), Pb(II) ad Zn(II).
Inorganic Chemistry, 2006, 45(14): 5510-5520
05 - Bonnot F, Molle T, Ménage S, Moreau Y, Duval S, Favaudon V, Houee-Levin C and Nivière V
Control of the evolution of iron peroxide intermediate in superoxide reductase from Desulfoarculus baarsii. Involvement of lysine 48 in protonation.
Journal of the American Chemical Society, 2012, 134(11): 5120-5130
Top page