Published on 25 January 2019
Body text 1
 pDYNAMO
pDYNAMO
http://www.pdynamo.org
Protein visualization program MPV3D
Serge Crouzy, Silouane Gérin (JD Lafontaine, MK Sautriau, M Ortega, JF Legout, J Flocard)
The development of MPV3D was not intended to compete with very good programs VMD, PyMOL ... but rather to insist on missing or complex features in the software and yet very useful in the early stages of modeling. For example:
- Quick Selection tool
- Flexibility in managing the display of a large number of molecules (pdb)
- Structure superposition
- Clear and quick display of labels on atoms and residues
- Interfacing to CHARMM program
Several students have contributed to this program during their internship (JD Lafontaine, MK Sautriau, Mr. Ortega, JF Legout, J Flocard ...)
The program has been recently ported to Qt4 for Linux systems in 2012 (Figure)
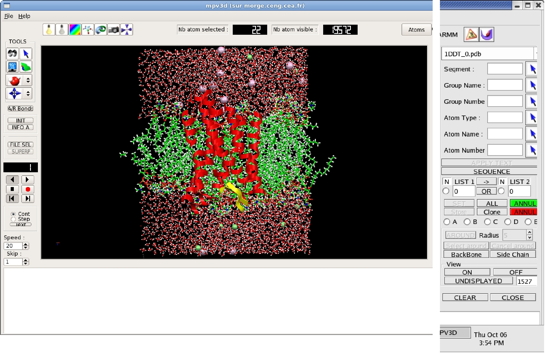
I-Introducing the main window and the selection window of the MPV3D program. The image shows a simulation of a G protein receptor inserted into an all-atom model DPPC phospholipid membrane including solvent and counterions.
Development of open access utility programs
Serge Crouzy et al.
We have developed a range of tools for analysis and visualization, most of them related to the molecular dynamics program CHARMM. These programs are freely available for academic users by simple request too Serge Crouzy.
 Multibox: to create water boxes (pdb or crd) of different shapes
Multibox: to create water boxes (pdb or crd) of different shapes
IR2D: Calculation of two-dimensional spectra from a set of 1D spectra recorded during the kinetics of folding of a protein, for example, using double Fourier transform. One of the two spectra allows the detection of correlated structural changes during the reaction and the other spectrum enables the detection of asynchronous changes (Noda I. 1998 Appl . Spectroscopy 47, 1329-1336 ).
Shell scripts:
Stride2gnu.sh, stride2gnu.com and stride.gnu: Create a graph of the evolution of the secondary structure of a protein from a set of PDBs
CHARMM input scripts:
Addions.inp: adds counterions to a protein structure by placing them at the minimum of the electrostatic potential calculated on a grid around the protein.
Membrane suite: a series of scripts to create a hydrated phospholipid bilayer and equilibrate a membrane protein within this bilayer, from ideas and scripts developed by T. Woolf and B. Roux.
AMRP: automatic map refinement protocol: script using CHARMM to calculate an adiabatic energy profile along a reaction coordinate
Wham: program for unbiasing free energy profiles obtained with the PERT command from CHARMM (from a list of energy values ) or from time series of one or two reaction coordinates constrained by the technique of "umbrella sampling" (based on the ideas of B. Roux).
Mpv3d: protein visualization program interfaced with CHARMM - Under development.
Top page