Reactivity of metalloproteins
Published on 25 January 2019
Body text 1
 Reactivity of « spore Photoproduct Lyase »
Reactivity of « spore Photoproduct Lyase »
Yohann Moreau, (Collaboration LCBM/BioCat and Inac)
The resistance to UV-induced damages of DNA of some bacterial spores is based on the activity of the iron-sulfur enzyme ''spore photoproduct lyase ” (SPL). Its role is to catalyze the repair mechanism of UV-induced thymin dimers. Experimental and theoretical studies have shown that the mechanism involved the radical 5'-deoxy-adenosyl. Recent work in the laboratory have shown that a particular cysteine plays a crucial role in the reaction process. The recent publication (2012) of the X-Ray structure of SPL in interaction with a portion of DNA allows us to consider the theoretical study of the repair mechanism, based on a realistic model which takes into account all the chemical environment involved. To achieve this goal, we make use of a multi-scale methodology in which the reactive part is described by quantum chemistry while the whole environment is modelled by molecular mechanics (QM / MM). This work is done within the framework of ANR funding "Sporepair" and therefore strong interaction with all partners involved in the project (experimentalists and theoretician chemist of the Laboratoire des lésions des acides nucléiques).
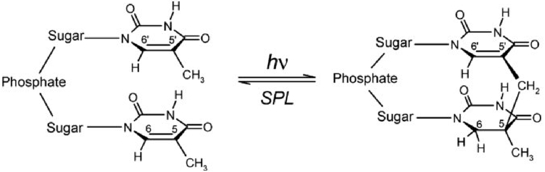
Reparation of UV-induced DNA damage by SPL.
Study of Superoxide Reductase
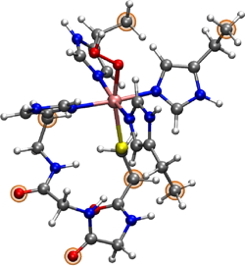
Superoxide reductase (SOR) is a non-heme iron metalloenzyme that detoxifies superoxide radical in microorganisms. Its active site consists of an unusual nonheme Fe(II) center in a [His4 Cys1 ] square pyramidal pentacoordination, with the axial cysteine ligand proposed to be an essential feature in catalysis.
|  |
| |
| QM model of the active site of SOR, binding hydroperoxyde |
Superoxide reductase (SOR) is a non-heme iron metalloenzyme that detoxifies superoxide radical in microorganisms. Its active site consists of an unusual nonheme Fe(II) center in a [His4 Cys1 ] square pyramidal pentacoordination, with the axial cysteine ligand proposed to be an essential feature in catalysis. By the mean of our theoretical work, in tight collaboration with V. Nivière in team BioCat at LCBM [1], we wish to deepen our understanding of the mechanisms taking place along the catalytic cycle. We have recently focused our work on the influence of the second coordination-sphere of the metal since experimental evidences have shown that the disruption of one hydrogen bond toward the cysteinate ligand could dramatically influence the catalytic mechanism and even lead to a different reactivity. This work, published in the Journal of BioInorganic Chemistry, makes use of small quantum models that include two, one or no H-bond with the cysteinate ligand. Using our models, we have shown how the hydrogen bonds of the second sphere of coordination could influence the behavior of the active site, particularly by modulating the well-known “push-effect” that occurs between sulfur iron and oxygenated substrates. [2] We are now using the QM/MM approach in order to further explore the properties of SOR, especially how the proteic environment influences the activity of this enzyme.
Modelling of the reactivity of an artificial metallo-enzyme: NikA-Ox
 | |
| |
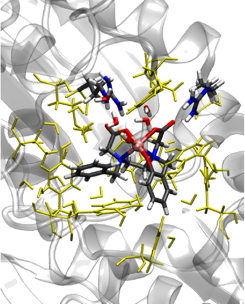
QM/MM model of the active site of NikA-Ox | |
The design of catalytic systems compatible with the principles of green chemistry is now an important issue. Using a bio-inspired approach, it is thus possible to create new species with tightly controlled properties. One can thus think about designing systems able to catalyze an enantio-selective reaction. One of the major activities of team BioCe at LCBM is the design and improvement of so-called artificial métalloenzymes. Inspired by iron mono-oxygenases enzymes, this team has set up a hybrid catalyst in which an inorganic complex is hosted by a protein (NikA). Experimental evidences (especially in cristallo reactivity) have shown the ability of this system to catalyze the double self arene-oxidation of the inorganic complex.
In order to understand the different steps leading to the formation of the cathecol, we have modeled the whole NikA-Ox system by the mean of a QM/MM approach. Starting from the Fe(IIII)-OOH structure found experimentally, we have considered two possible mechanisms leading to the phenolic intermediate. The first involves the formation of a hydroxide radical that adds directly to the phenyl ring, like in the first step of the degradation of heme-oxygenase. The second reactive path involves one further protonation of the distal oxygen, which leads to the formation of a (formally) Fe(V)=O intermediate, followed by the addition of the proximal oxygen atom. The first results indicate that the second mechanism is the most probable.
01 - Bonnot F, Molle T, Ménage S, Moreau Y, Duval S, Favaudon V, Houee-Levin C and Nivière V
Control of the evolution of iron peroxide intermediate in superoxide reductase from Desulfoarculus baarsii. Involvement of lysine 48 in protonation.
Journal of the American Chemical Society, 2012, 134(11): 5120-5130
01 - Tremey E, Bonnot F, Moreau Y, Berthomieu C, Desbois A, Favaudon V, Blondin G, Houee-Levin C and Nivière V
Hydrogen bonding to the cysteine ligand of superoxide reductase: Acid-base control of the reaction intermediates.
Journal of Biological Inorganic Chemistry, 2013, 18(7): 815-830
Top page