Les amyloses ont traditionnellement été définies comme des maladies dans lesquelles des protéines normalement solubles s'accumulent dans l'espace extracellulaire des tissus sous forme de dépôts insolubles de fibres riches en structures « feuillet-ß ». Celles-ci incluent des petites protéines solubles comme dans les pathologies neurodégénératives (maladie d'Alzheimer, maladie de Parkinson…), ou des grosses protéines pour les amyloses périphériques (amyloïdose systémique principal, diabète pancréatique, neuropathie du système nerveux autonome…). Dans le cas des maladies neurodégénératives, la formation des dépôts amyloïdes est associée à un stress oxydatif. L'étiologie des amyloses dégénératives montre des mécanismes cellulaires et moléculaires identiques ; l'apparition de dépôts amyloïdes est associée à une apoptose anormale.
Nous avons montré que Bcl-xL, une protéine de la famille Bcl-2 impliquée dans la régulation de l'apoptose, forme des fibres amyloïdes in vitro, mais aussi dans des cellules issues d'un neuroblastome dans certaines conditions de stress oxydant (Figure 1). Ces résultats suggèrent un lien possible entre apparition de dépôts amyloïdes et apoptose anormale. En effet, Bcl-xL étant une protéine anti-apoptotique, son implication dans des dépôts amyloïdes, devrait entrainer son inactivation et, en conséquence, une augmentation de l'apoptose (Figure 1). Nous explorons cette possibilité par des approches allant de la biophysique à la biologie cellulaire.
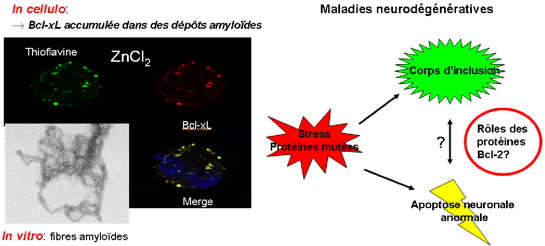
Figure 1 : (Microscopie de fluorescence) Immunolocalisation de dépôts amyloïdes (marquage thioflavine T, vert), de Bcl-xL (anticorps marqué avec un Alexa-647, marquage rouge) dans des cellules SHSY5Y en apoptose induite par ZnCl2. (Microscopie électronique) fibres amyloïdes formées par Bcl-xL in vitro.