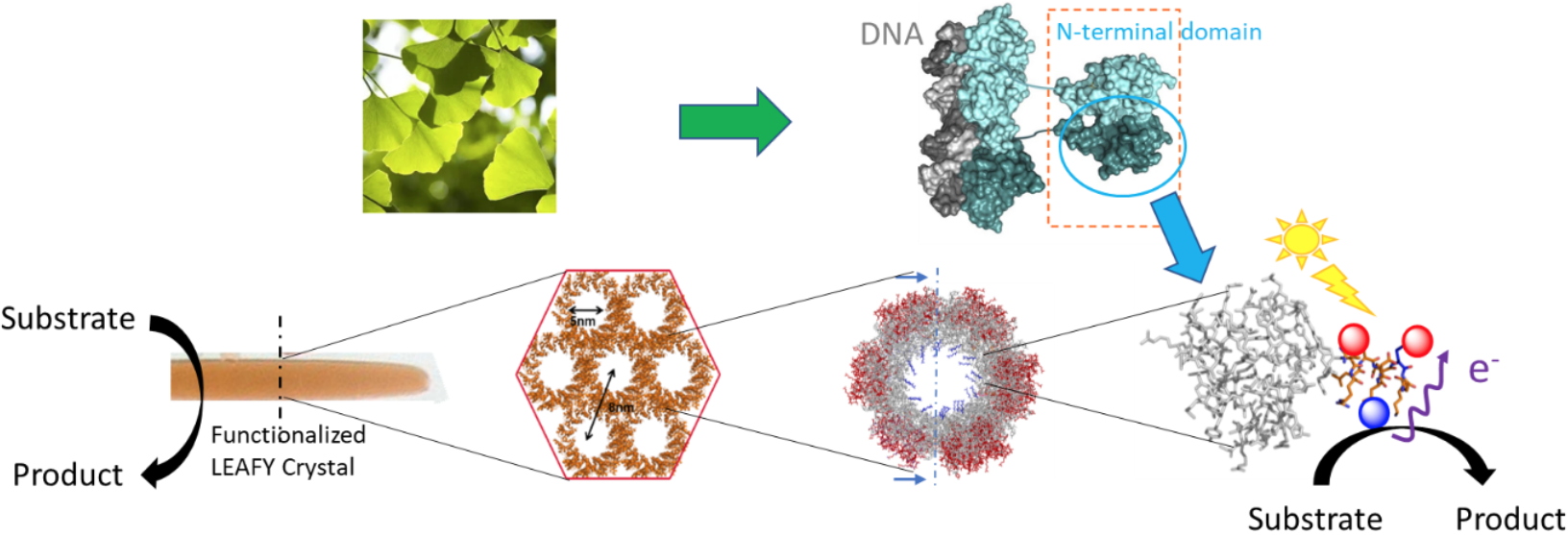
Les assemblages organisés de protéines peuvent présenter de nombreuses propriétés qui justifient leur utilisation pour la conception de bionanomatériaux innovants. Au laboratoire, en collaboration avec Renaud Dumas (LPCV), des cristaux du domaine d'oligomérisation de la protéine LEAFY du
, présentant une architecture en nid d'abeille, sont utilisés comme plateforme modulaire pour le greffage sélectif de différents complexes et nanoparticules pour effectuer des réactions données et accéder à des matériaux biohybrides fonctionnels innovants. Très récemment, des complexes de ruthénium ont été sélectivement greffés au sein de la structure cristalline pour effectuer de la photocatalyse d’oxydation asymétrique. Le matériau bio-hybride résultant a été entièrement caractérisé par spectroscopie de résonance Raman et UV-visible ainsi que par spectrométrie de masse et LC-MS après digestion enzymatique sélective. Il a été intéressant d’observer que l'insertion de complexes dans la structure tubulaire a permis une augmentation impressionnante de la stabilité des cristaux, évitant le recours à des stratégies de réticulation pour une utilisation future en tant que biocatalyseur hétérogène. Nous travaillons également sur l’incorporation sélective et contrôlée de nanoparticules au sein de cristaux obtenus par auto-assemblages de différents mutants dans le but d’effectuer des réactions d’hydrogénation via la réduction de protons. L’objectif ultime étant de développer des microréacteurs montés pour effectuer de la catalyse tandem. Cette activité soutenue par le programme DRF-Impulsion du CEA ainsi que par le programme MITI 80’ du CNRS est une collaboration avec R. Dumas (
) et P.-H. Elchinger (
).
 Dégradation-recyclage enzymatique et chimique des plastiques poyoléfiniques de type polyéthylène et polypropylène
Dégradation-recyclage enzymatique et chimique des plastiques poyoléfiniques de type polyéthylène et polypropylène
Collaboration V. Nivière, LCBM Avec les changements climatiques, la pollution par les plastiques est un problème majeur auquel nous devons faire face. Plus de 348 millions de tonnes de déchets plastiques ont été générées en 2017, et un tiers s’est retrouvé dans la nature avec des conséquences dramatiques sur les écosystèmes. Parmi les différents types de plastiques, les polyoléfines comme le polyéthylène PE (sacs plastiques, bouteilles…) et le polypropylène PP (emballages rigides) sont particulièrement préoccupants car ils constituent la majeure partie des déchets plastiques que l’on retrouve à la surface de la terre. Cela est lié d’une part à leur production industrielle très importante mais également à leur très grande stabilité vis-à-vis de la dégradation en raison de leurs structures chimiques particulièrement résistantes ne présentant que des liaisons fortes C-C et C-H. La connaissance des mécanismes par lesquels ces plastiques peuvent être dégradés, en particulier par les microorganismes, représente donc un enjeu de toute première importance.
La dégradation des plastiques en milieu naturel implique deux phases. Elle est généralement initiée par une phase abiotique pendant laquelle des premières modifications chimiques apparaissent sous l’effet du rayonnement solaire et d’actions mécaniques. Ensuite, une phase biotique prend le relais, durant laquelle les microorganismes interviennent en utilisant notamment les défauts du plastique présents soit dans le polymère d’origine soit générés après la première phase. Malheureusement, ces processus biotiques sont souvent peu efficaces surtout en ce qui concerne le PE. Dans ce dernier cas, un certain nombre de bactéries et de champignons ont été identifiées comme pouvant être impliquées dans sa biodégradation, mais avec cependant des croissances cellulaires très lentes et des quantités de PE dégradé au final assez limitées. La structure du PE, avec des liaisons chimiquement très stables associée à une surface complètement hydrophobe limitant la fixation des organismes, pose effectivement la question des mécanismes par lesquels des enzymes peuvent agir efficacement dessus. Actuellement rien n’est connu sur ces mécanismes de biodégradation. Il semble néanmoins que des enzymes de type peroxydases ou laccases sécrétées par les microorganismes, permettent l’oxydation des liaisons C-C ou C-H et/ou les défauts générés durant la phase abiotique. Ces oxydations enzymatiques permettraient une fragmentation du PE, générant de plus petits morceaux fonctionnalisés (donc plus solubles), alors directement assimilables par les microorganismes. Ainsi, avec V. Nivière (LCBM,
équipe Biocat) nous avons ciblé ce type d’enzymes afin d’évaluer leur efficacité lors de la dégradation des dérivés polyoléfiniques avant et après traitement abiotique et d’étudier également l’importance de différents médiateurs rédox dans le processus de dégradation.
 Collaborations
Collaborations
 La quinolinate synthase. Une cible interessante pour lutter contre H. Pylori,et M. Leprae. Synthèse d’inhibiteurs et étude de leur mécanismes d’inhibition
La quinolinate synthase. Une cible interessante pour lutter contre H. Pylori,et M. Leprae. Synthèse d’inhibiteurs et étude de leur mécanismes d’inhibition
Collaboration S. Ollagnier, LCBM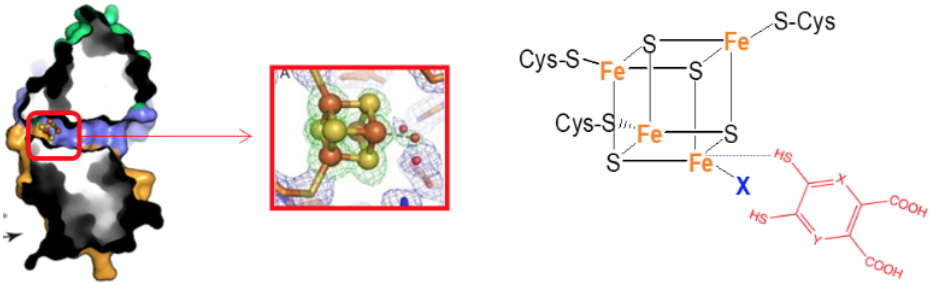
La quinolinate synthase est une enzyme impliquée dans la synthèse de l’acide quinolinique, chez
H. pylori et
M. leprae. Ces bactéries ne possédant pas de voie de secours pour la production de ce métabolite essentiel sont de ce fait des cibles particulièrement intéressantes. Depuis quelques années, en collaboration avec le Dr S. Ollagnier (LCBM,
équipe Biocat), nous travaillons sur le développement d’inhibiteurs de cette enzyme ce qui nous a permis de découvrir une famille de molécules présentant des activités inhibitrices intéressantes. Très récemment, un certain nombre de membres de cette famille ont été synthétisés permettant de réaliser une étude complète du mécanisme d’action.
Design of specific inhibitors of quinolinate synthase based on [4Fe–4S] cluster coordination J. Saez Cabodevilla, A. Volbeda, O. Hamelin, J.-M. Latour, O. Gigarel, M. Clemancey, C. Darnault, D. Reichmann, P. Amara, J. C. Fontecilla-Camps, S. Ollagnier de Choudens,
Chem. Commun.,
2019, 55, 3725 Crystallographic Trapping of Reaction Intermediates in Quinolinic Acid Synthesis by NadA. A. Volbeda, J. Saez Cabodevilla, C. Darnault, O. Gigarel, T.-H.-L. Han, O. Renoux, O. Hamelin, S. Ollagnier-de-Choudens, P. Amara, J. C. Fontecilla-Camps.
ACS Chem. Biol.,
2018, 13, 1209 Studies of Inhibitor Binding to the [4Fe-4S] Cluster of Quinolinate Synthase. A; Chan, M. Clémancey, J.-M. Mouesca, P. Amara, O. Hamelin, J.-M. Latour, S. Ollagnier de Choudens, Angew. Chem. Int. Ed. ,
2012, 51, 7711Synthèse de molécules marquées pour l’étude structurale et dynamique de macroprotéines par RMN - De la recherche vers la création
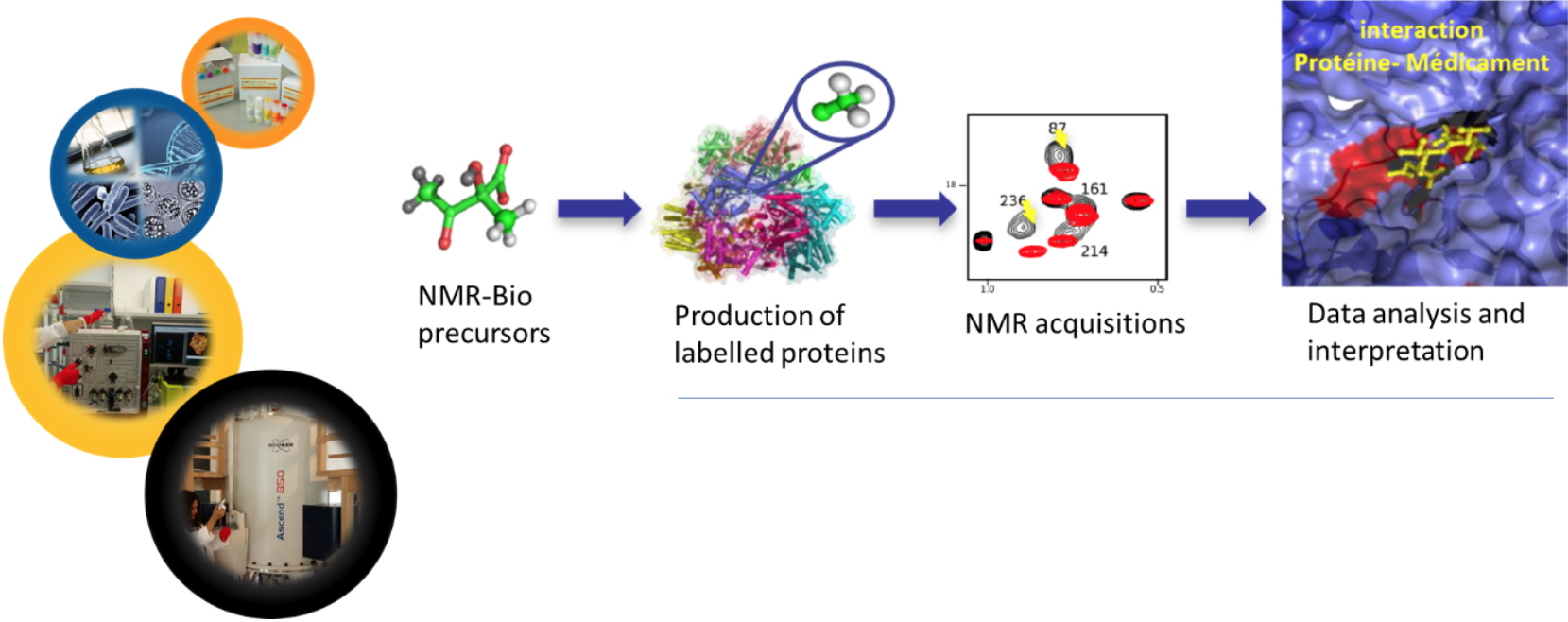
Collaboration J. Boisbouvier (IBS), E. Crublet (NMR-Bio), R. Kerfah (NMR-Bio) La RMN est une méthode de choix pour étudier les caractéristiques structurales et dynamiques des biomacromolécules en solution aqueuse. Cependant cette technique se caractérise par une faible sensibilité et est limitée à des objets biologiques de tailles modestes. L’équipe de J. Boisbouvier avec qui je collabore depuis 2009, avait montré que le marquage spécifique (
2H,
13C) de macroprotéines permettait de repousser les limites de cette technique pour observer des objets de plus en plus gros en un temps expérimental de plus en plus court. Nous avons montré que des dérivés sélectivement marqués de l’acétolactate pouvaient être incorporés dans les voies de biosynthèse des leucine, Valine et Isoleucine (
Angew. Chem. Int Ed.2010). Deux brevets ont été déposés en 2010 (PCT/IB2010/000282) et 2012. Cette collaboration a conduit à la création de la société
NMR-Bio en janvier 2016. En parallèle aux activités de valorisation, nous nous concentrons sur le développement et la production chimique et enzymatique des autres acides aminés méthylés (alanine, méthionine, et thréonine) sélectivement marquées ainsi que leur incorporation dans des protéine d’intérêt en évitant les fuites métaboliques.
Asymmetric Synthesis of Methyl Specifically Labelled L Threonine and Application to the NMR Studies of High Molecular Weight Proteins, I. Ayala, L. Chiari, R. Kerfah, J. Boisbouvier,* P. Gans, O. Hamelin*,
Chem. Select.,
2020, 5, 1– 7 CH3-specific NMR Assignment of Isoleucine, Leucine, Valine Methyl Groups in High Molecular Weight Proteins. R. Kerfah, O. Hamelin, J. Boisbouvier, D. Marion,
J. Biol. NMR,
2015, 63, 389 Scrambling free combinatorial labeling of alanine-b, isoleucine-d1, leucine-proS and valine-roS methyl groups for the detection of long range NOEs. R. Kerfah, , M. J. Plevin, O. Pessey, O. Hamelin, P. Gans, J. Boisbouvier,
J. Biol. NMR,
2015, 61, 73 Specific labelling and assignment strategies of valine methyl groups for NMR studies of high molecular weight proteins. G. Mas, E. Crublet, O. Hamelin, P. Gans, J. Boisbouvier,
J. Biol. NMR,
2013, 57, 251 An optimized isotopic labelling strategy of isoleucine-?(2) methyl groups for solution NMR studies of high molecular weight proteins. I. Ayala, O. Hamelin, C. Amero, O. Pessey, M. J. Plevin, P. Gans, J. Boisbouvier,
Chem. Commun.,
2012, 48, 1434 A simple biosynthetic method for stereospecific resonance assignment of prochiral methyl groups in proteins. M. J. Plevin, O. Hamelin, J. Boisbouvier, P. Gans,
J. Biol. NMR,
2011, 49, 61 Stereospecific Isotopic Labeling of Methyl Groups for NMR Spectroscopic Studies of High-Molecular-Weight Proteins. P. Gans, O. Hamelin, R. Sounier, I. Ayala, M.A. Durà, C. D. Amero, M. Noirclerc-Savoye, B. Franzetti, M. J. Plevin, J. Boisbouvier.
Angew. Chem. Int. Ed.,
2010, 49, 1958NMR-Bio (http://www.nmr-bio.com)

La société
NMR-Bio est une organisation de recherche contractuelle qui est née d'une collaboration entre l'Institut de biologie structurelle (IBS) et le Laboratoire de chimie et de biologie des métaux (LCBM).
NMR-Bio fournit des précurseurs pour le marquage sélectif des groupes méthyle dans les protéines perdeutérées ainsi qu'une large gamme de services pour les applications de résonance magnétique nucléaire (RMN) et de microscopie électronique (EM). En outre,
NMR-Bio investit fortement dans la R&D pour développer des stratégies et des solutions innovantes afin d'aider ses clients à surmonter leurs problèmes techniques. En collaboration avec ses partenaires universitaires,
NMR-Bio mène un programme de recherche scientifique stimulant qui vise à étendre l'approche de marquage sélectif, une stratégie pertinente pour l'étude RMN des machineries supramoléculaires et qui est appliquée de façon routinière dans
E. coli, aux cellules de mammifères, un des systèmes d'expression les plus demandés par l'industrie pharmaceutique. Des protocoles visant à obtenir ce marquage dans les cellules d'insectes et les systèmes sans cellules ont déjà été mis en place au cours des deux premières années de cette collaboration. Je suis un des cofondateurs de la société issue des travaux de recherche réalisés en collaboration avec J. Boisbouvier (Institut de Biologie Structurale de Grenoble). En tant que consultant scientifique de 2016 à 2020 j’étais en charge de l'élaboration de nouvelles stratégies de synthèse d'acides aminés marqués et de précurseurs.