Ruthenium complexes have innumerable properties, including for example the ability to exist in enantiomeric forms, to be oxidizing species (especially in their Ru(IV)=O form) and also to have photophysical properties that can make them particularly attractive chromophores. Over the last 20 years, these different properties have been exploited and combined with one main goal: To carry out (asymmetric) (photo)catalysis which is gradually becoming part of a green chemistry.
Usually chiral catalysts are formed by a metal cation (usually, Mn, Cu, Ti…) surrounded by an organic chiral ligand responsible of the asymmetric induction. Few years ago, using ruthenium based complexes, because of their well known relative configurational stability, we demonstrated that chiral mononuclear complexes, in which the only stereogenic center was the metal ion itself (chirality Δ, Λ for octahedral complexes), were able to catalyze asymmetric oxidations. This study showed that a crystallization-induced asymmetric transformation allows to obtain ruthenium complexes with an octahedral geometry with enantiomeric excesses of up to 98%..
Since, an original evolution of our research program was to design “chiral-at-metal” complexes so that they display free coordinating moieties (named chiral metalloligand) for binding a second metal ion on which the reaction would take place. Asymmetric catalytic transfer hydrogenation was then carried out with an original dinuclear ruthenium complex constituted by the chiral metalloligand and a Ru(p-cymene)Cl moiety acting as a catalytic center. Even if the results, in both terms of catalytic activity and enantioselectivity did not allowed to rival with the most efficient asymmetric transfer hydrogenation catalysts developed to date bearing chiral organic ligands, this novel class of chiral catalysts, based on a chiral-at-metal metalloligand has opened a new research direction.
Taking into account the absolute necessity to develop an ever cleaner and economical chemistry, it was envisaged to develop new “eco-aware” methods for catalytic oxidation. Inspired by the photosystem we developed two Ru-based dyads able to perform sulfide oxidation by activation of water as unique source of oxygen atom. These dyads combine covalently a photosensitizer capable of harvesting light and a catalytic fragment. Mechanistic studies have highlighted the involvement of an electron transfer process that can be coupled with proton transfer leading to the formation of a Ru(IV)=O oxidative species.
Thanks to common effort between O. Hamelin and S. Torelli, supported by the Arcane labex we demonstrated that the covalent combination of a ruthenium-based photosensitizer with a copper-based catalytic partner allows the efficient oxygenation of various substrates by photoactivation of O2 as the unique oxygen atom source. Photophysical studies highlighted a photo-induced electron transfer from the chromophore to the catalyst upon light irradiation. The efficiency of the dyad compared to bimolecular conditions (
) also emphasized a synergistic effect.
In our research program aiming to develop new “eco-aware” catalysts, we were interested in the covalent combination of a photosensitizer and a catalyst to achieve catalytic light-driven oxidation of organic substrates using water as the unique oxygen atom source. This approach avoids the use of classical (and sometimes relatively toxic and/or hazardous) oxidants. However, as most of the photocatalytic systems reported in the literature for oxidation or reduction reactions, a sacrificial reagent is used to either deliver or accept electrons resulting of the reaction. This “electrons-waste” is also accompanied by the generation of by-products issued from the sacrificial chemical. In order to prevent this drawback, it was envisaged to associate to the oxidative photocatalytic system a second catalyst able to add value to the liberated electrons. Therefore, it was envisaged to synthesize a multi-component system combining a ruthenium-based catalyst able to activate water, a copper-based one for dioxygen activation and using the electrons generated by the first catalyst, and a photosensitizer allowing the sequential electron transfer from one catalyst to the other. This project was supported by an ANR funding (TROICAT).
 Development of biohybrids materials for catalysis
Development of biohybrids materials for catalysis
Collaboration R. Dumas (Irig-LPCV) et P.-H. Elchinger (Irig-SyMMES)
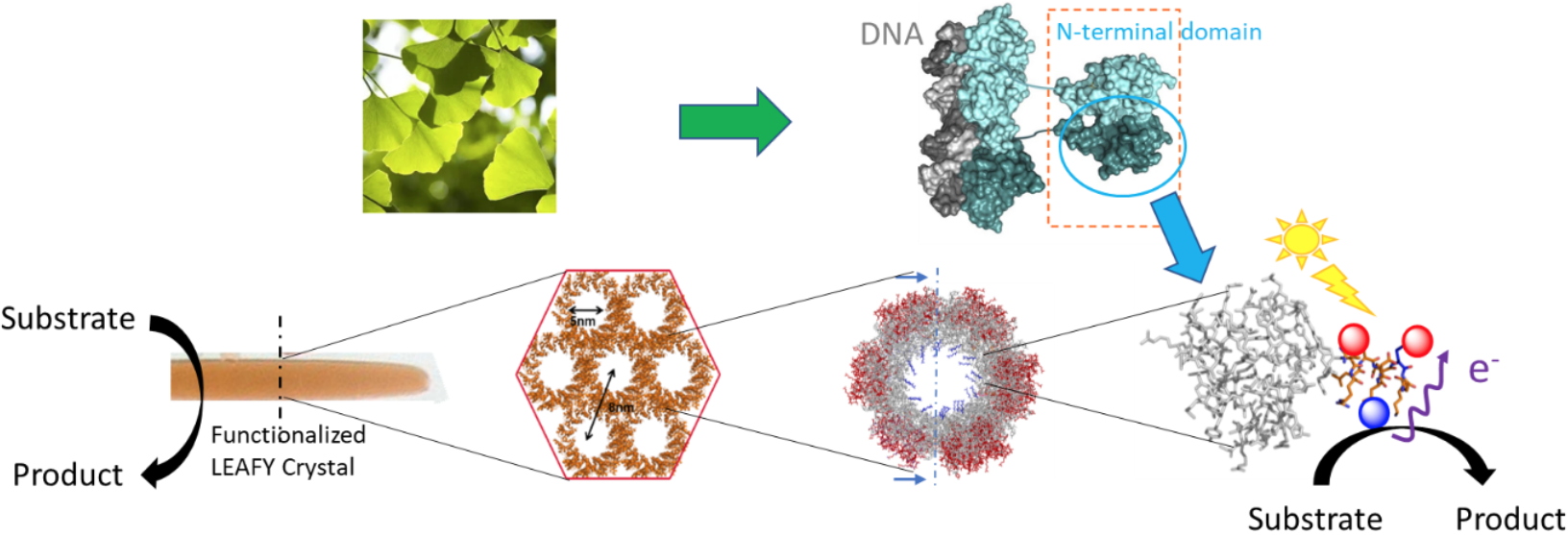
Well-organized protein assemblies offer many properties that justify their use for the design of innovative bionanomaterials. In collaboration with R. Dumas (
LPCV), crystals of the oligomerization domain of the LEAFY protein from
Ginkgo biloba, organized in a honeycomb architecture, are used as a modular platform for the conception of functional bio-hybrid materials. Very recently, ruthenium complexes were selectively grafted within the crystalline material to perform heterogeneous photocatalytic oxidation. The resulting bio-hybrid material was fully characterized by UV-visible and resonance Raman spectroscopies as well as by mass spectrometry and LC-MS after selective enzymatic digestion. Interestingly, insertion of such complexes within the tubular structure affords an impressive increase in stability of the crystals avoiding the use of stabilizing cross-linking strategies. We are also working on the selective and controlled incorporation of nanoparticles into crystals obtained by self-assembly of different mutants in order to carry out hydrogenation reactions via proton reduction.
The ultimate goal is to develop microreactors mounted for tandem catalysis. This activity, supported by the CEA's DRF-Impulse programme and the CNRS's MITI 80' programme, is a collaboration with R. Dumas (
LPCV) and P.-H. Elchinger (
SyMMES).
LEAFY protein crystals with a honeycomb structure as plateform for selective preparation of stable bio-hybrid materials. L. Chiari, P. Carpentier, S. Kieffer-Jaquinod, A. Gogny, J. Perard, S. Ravanel, S. Ménage, R. Dumas, O. Hamelin, submitted to Chem. Bio. Chem.
 Biochemical and chemical degradation of polyolefinic plastics such as polyethylene and polypropylene
Biochemical and chemical degradation of polyolefinic plastics such as polyethylene and polypropylene
Collaboration V. Nivière, LCBM
Along with climate change, pollution from plastics is a major problem we have to face. More than 348 million tons of plastic waste were generated in 2017, and a third of this was released into the environment with dramatic consequences for ecosystems. Among the different types of plastics, polyolefinic plastics such as polyethylene PE (plastic bags, bottles...) and polypropylene PP (rigid packaging) are of particular concern as they make up the bulk of the plastic waste found on the earth's surface. This is related on the one hand to their very high industrial production but also to their very high stability against abiotic and biotic degradations due to their particularly resistant chemical structures with only strong C-C and C-H bonds in addition to a completely hydrophobic surface which makes the fixation of microorganisms particularly difficult. The characterization of the mechanisms by which these plastics can be degraded, particularly by microorganisms, is therefore a challenge of the utmost importance. However, even if such plastics are considered to be practically inert, recent data showed that a number of bacteria are involved in their biodegradation. But, currently, nothing is known about the biodegradation mechanisms. It seems nevertheless that enzymes such as peroxidase or laccases, secreted by the microorganisms, allow the oxidation of the C-C or C-H bonds and/or the defects generated during the abiotic phase. These enzymatic oxidations would allow the fragmentation of PE into smaller functionalized molecules (therefore more soluble) directly assimilable by the microorganisms. Thus, with V. Nivière from
Biocat team, we have targeted this type of enzymes in order to evaluate their efficiency during the degradation of polyolefin derivatives before and after abiotic treatment and decided to study the importance of different redox mediators in the degradation process.
Collaborations
 Design of inhibitors as potential antibacterial agents and investigation of their inhibition mechanisms
Design of inhibitors as potential antibacterial agents and investigation of their inhibition mechanisms
Collaboration S. Ollagnier, LCBM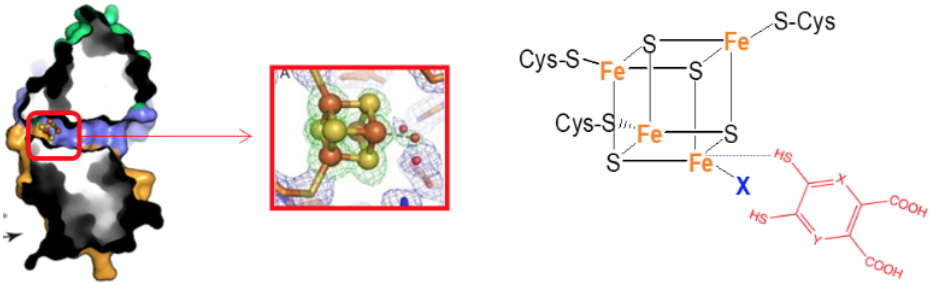
Biosynthesis of nicotinamide adenine dinucleotide (NAD), an essential and ubiquitous cofactor in biology, involves in all living organisms the synthesis of quinolinic acid (QA). In mammals and some bacteria, QA can be synthesized from L-tryptophan whereas in most prokaryotes it is generated from L-aspartate and dihydroxyacetone phosphate (DHAP) through the concerted action of two enzymes: the L-aspartate oxidase NadB that first converts L-aspartate to iminoaspartate (IA), and the quinolinate synthase NadA which condenses IA with DHAP to form QA. Besides these de novo pathways for NAD synthesis, salvage pathways exist in some organisms allowing the coenzyme to be recycled from small metabolites such as niacin or nicotinamide. The different pathways for QA biosynthesis in most prokaryotes and eukaryotes, combined with the fact that salvage pathways are absent in some pathogenicmicroorganisms such as
Mycobacterium leprae and
Helicobacter pylori, makes NadA an ideal target in the search for specific antibacterial drugs. All NadA proteins bind an oxygen-sensitive [4Fe–4S] cluster that is essential for its activity. The cluster is ligated by three strictly conserved cysteines and an exchangeable water molecule. In 2012, we demonstrated that 4,5-dithiohydroxyphthalic acid (DTHPA), a structural analogue of QA, binds irreversibly to the [4Fe–4S] cluster of
Escherichia coli NadA. Binding of DTHPA inhibits EcNadA activity, which strongly suggested that the unique iron site of the [4Fe–4S] cluster was intimately involved in QA synthesis. This was unambiguously demonstrated by crystal structures of
Thermotoga maritima [4Fe–4S]–NadA active site mutants in which intermediates bound to the accessible iron site were trapped. Very recently, we reported the synthesis and activity of derivatives of the first inhibitor of NadA (DTHPA). Using multidisciplinary approaches we have investigated their action mechanism and discovered additional specific inhibitors of this enzyme.
Design of specific inhibitors of quinolinate synthase based on [4Fe–4S] cluster coordination J. Saez Cabodevilla, A. Volbeda, O. Hamelin, J.-M. Latour, O. Gigarel, M. Clemancey, C. Darnault, D. Reichmann, P. Amara, J. C. Fontecilla-Camps, S. Ollagnier de Choudens, Chem. Commun., 2019, 55, 3725
Crystallographic Trapping of Reaction Intermediates in Quinolinic Acid Synthesis by NadA. A. Volbeda, J. Saez Cabodevilla, C. Darnault, O. Gigarel, T.-H.-L. Han, O. Renoux, O. Hamelin, S. Ollagnier-de-Choudens, P. Amara, J. C. Fontecilla-Camps. ACS Chem. Biol., 2018, 13, 1209
Studies of Inhibitor Binding to the [4Fe-4S] Cluster of Quinolinate Synthase. A; Chan, M. Clémancey, J.-M. Mouesca, P. Amara, O. Hamelin, J.-M. Latour, S. Ollagnier de Choudens, Angew. Chem. Int. Ed. , 2012, 51, 7711
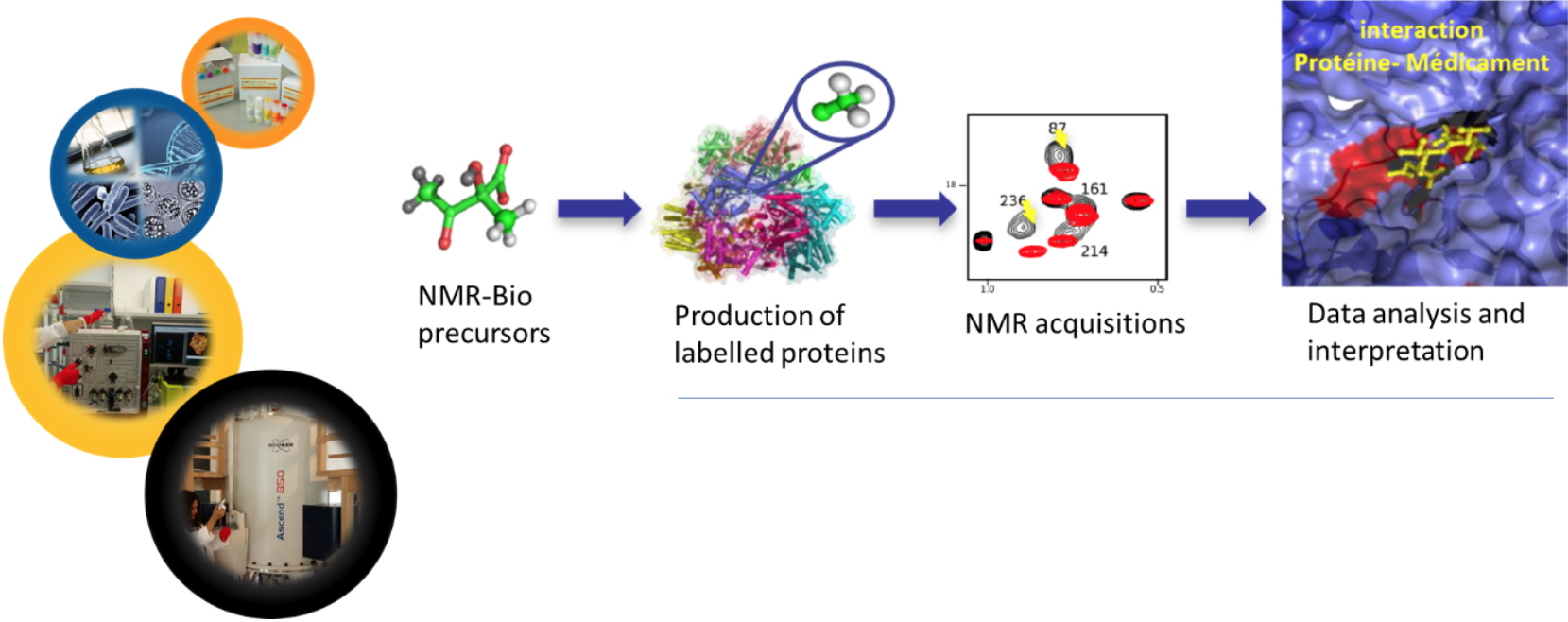
Synthesis of labeled molecules for the structural and dynamic study of macroproteins by NMR- From research to creation
Collaboration J. Boisbouvier (IBS), E. Crublet (NMR-Bio), R. Kerfah (NMR-Bio)
NMR is a method of choice for studying the structural and dynamic characteristics of biomacromolecules in aqueous solution. However, this technique is characterized by low sensitivity and is limited to small biological objects. J. Boisbouvier's team, with whom I have been collaborating since 2009, had shown that the specific labeling (
2H,
13C) of macroproteins allowed to push back the limits of this technique to observe larger objects in a smaller experimental time. We showed that selectively labelled acetolactate derivatives could be incorporated into the leucine, Valine and Isoleucine biosynthetic pathways. Two patents were filed in 2010 (PCT/IB2010/000282) and 2012. This collaboration led to the creation of the company
NMR-Bio in January 2016. In parallel to the valorisation activities, we focused on the development and chemical and enzymatic production of other selectively labelled methylated amino acids (alanine, methionine, and threonine) as well as their incorporation into proteins of interest while avoiding metabolic leaks.
Asymmetric Synthesis of Methyl Specifically Labelled L Threonine and Application to the NMR Studies of High Molecular Weight Proteins, I. Ayala, L. Chiari, R. Kerfah, J. Boisbouvier,* P. Gans, O. Hamelin*, Chem. Select., 2020, 5, 1– 7
CH3-specific NMR Assignment of Isoleucine, Leucine, Valine Methyl Groups in High Molecular Weight Proteins. R. Kerfah, O. Hamelin, J. Boisbouvier, D. Marion, J. Biol. NMR, 2015, 63, 389
Scrambling free combinatorial labeling of alanine-b, isoleucine-d1, leucine-proS and valine-roS methyl groups for the detection of long range NOEs. R. Kerfah, , M. J. Plevin, O. Pessey, O. Hamelin, P. Gans, J. Boisbouvier, J. Biol. NMR, 2015, 61, 73
Specific labelling and assignment strategies of valine methyl groups for NMR studies of high molecular weight proteins. G. Mas, E. Crublet, O. Hamelin, P. Gans, J. Boisbouvier, J. Biol. NMR, 2013, 57, 251
An optimized isotopic labelling strategy of isoleucine-?(2) methyl groups for solution NMR studies of high molecular weight proteins. I. Ayala, O. Hamelin, C. Amero, O. Pessey, M. J. Plevin, P. Gans, J. Boisbouvier, Chem. Commun., 2012, 48, 1434
A simple biosynthetic method for stereospecific resonance assignment of prochiral methyl groups in proteins. M. J. Plevin, O. Hamelin, J. Boisbouvier, P. Gans, J. Biol. NMR, 2011, 49, 61
Stereospecific Isotopic Labeling of Methyl Groups for NMR Spectroscopic Studies of High-Molecular-Weight Proteins. P. Gans, O. Hamelin, R. Sounier, I. Ayala, M.A. Durà, C. D. Amero, M. Noirclerc-Savoye, B. Franzetti, M. J. Plevin, J. Boisbouvier. Angew. Chem. Int. Ed., 2010, 49, 1958
NMR-Bio (http://www.nmr-bio.com)

The Startup
NMR-Bio is a contract research organization that arose from a collaboration between the Structural Biology Institute (IBS) and the Laboratory of Chemistry and Biology of Metal.
NMR-Bio supplies precursors for the selective labelling of methyl groups in perdeuterated proteins along with a broad range of services for Nuclear Magnetic Resonance and Electron Microscopy applications. In addition, NMR-Bio invests heavily in the R&D to develop innovative strategies and solutions to help its customers to overcome their technical issues. In collaboration with its academic partners,
NMR-Bio is conducting a challenging scientific research program which aims to extend the selective labelling approach, a pertinent strategy to the NMR study of supramolecular machineries and which is routinely applied in
E. coli, to the mammalian cells, one of the most requested expression systems by the pharmaceutical industry. Protocols to achieve this labelling in insect cells and Cell-Free systems have been already set-up in the first two years of this collaboration. I am one of the co-founders of the company that emerged from the research work in collaboration with J. Boisbouvier. As scientific advisor from 2016 to 2020 I was involved in the development of new strategies for the synthesis of labeled amino acids and precursors.