Role of copper transporter proteins in chronic inflammation: The case of cystic fibrosis
Published on 6 October 2025
Body text 1
Mohamed Benharouga, associate professor at the Grenoble University (UJF)
 Research topics
Research topics
Chronic inflammatory diseases represent the third leading cause of death in France, just after cardiovascular diseases and cancers. Among these pathologies, it denotes a significant number of pulmonary disease. The acute respiratory distress syndrome, broncho chronic obstructive pulmonary disease, invasive aspergillosis, or cystic fibrosis are part of these pathologies. Cystic fibrosis is an autosomal recessive genetic disease most common in Caucasians. The gene encodes a transmembrane protein called CFTR (Figure) [Riordan et al., 1989]. Mutations in this gene lead to a deregulation of the chloride channel and Na+ associated with dehydration of the mucus, bronchial inflammation and infection leading to respiratory failure, obstructive type, the first cause of morbidity and mortality. The cascade of events associated with disease progression might involve first triggering lung inflammation and secondly, the appearance of a respiratory tract infection [Armstrong et al., 1997], leaving suggest that other events could be responsible for its installation. Oxidative stress, widely accepted as a critical component of many diseases, may be linked to the development of coronary heart disease, stroke, cancer and Alzheimer's disease. It is defined by an imbalance between production of reactive oxygen and nitrogen species (ROS) and defense mechanisms and detoxification. Thus, the actors of oxidative stress may have deleterious effects on many cell components such as proteins, lipids or nucleic acids
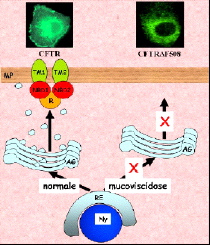 Figure : Biosynthesis and cellular expression of the wild and mutated ΔF508.
Figure : Biosynthesis and cellular expression of the wild and mutated ΔF508.
The CFTR protein (Cystic Fibrosis transmembrane regulator) has two membrane domains, each consisting of 6 transmembrane segments, and 3 cytoplasmic domains: 2 nucleotide-binding domains and a regulatory domain. CFTR forms an anion channel allowing diffusion of chloride ions after phosphorylation of the regulatory domain, MgATP binding and hydrolysis. Like all membrane proteins, CFTR is synthesized in the endoplasmic reticulum (ER). During its export to the plasma membrane, CFTR matures in the Golgi apparatus (GA) The most frequent mutation (70% of patients) is the deletion of the first domain nucleotide Phe (F508). This mutation causes the stopping of CFTR in the reticulum and its degradation by mechanisms still poorly understood. When one succeeds by a trick to correctly address to the plasma membrane, F508 forms a functional chloride channel. It is therefore very important to understand how F508 is degraded in the reticulum to establish a strategy to ensure its insertion into the plasma membrane of diseased cells and restoring chloride transport.
In cystic fibrosis, the importance of the phenomena of infection and inflammation contributes to the excess production of oxygen free radicals, which inadequately compensated by the antioxidant defenses would be involved in the impairment of lung function [Lagrange-Puget et al., 2004]. Observed in patients with a decreased activity of three enzymes involved in detoxification of the cell: superoxide dismutase dependent on copper and zinc (Cu / Zn-SOD), cytochrome C oxidase (copper-dependent) and glutathione peroxidase (GP) [Best et al., 2004]. There is also a deficiency in the secretion of lung glutathione (GSH) whose primary function is the elimination of toxic substances and neutralizing heavy metals such as copper (Cu) [Rottner et al., 2009]. Cu, the third transition element most abundant in the cells after iron and zinc is essential for life in eukaryotes and prokaryotes. Disruption of its metabolism, which may be genetic (Wilson, Menkes, ..) or acquired (oxidative stress causing chronic inflammation) [Coronado et al., 2005], can cause severe damage cell. Thus, Cu homeostasis is tightly regulated by the intervention of transporters such as P-type ATPases (ATP7A and ATP7B), and regulatory proteins such as prion protein (PrP), the protein COMMD1 and métallochaperonnes [Tao and Gitlin, 2003]. In case of inflammation, a copper treatment may induce an increase in the number of circulating neutrophils in the lung, IL-8 secretion via ROS production, IL-6, and activation of NFγB and COX2 [Bar-Or et al., 2003]. It follows from all these data that copper homeostasis in cystic fibrosis could be a determining factor in the initiation and maintenance of lung inflammation. Our studies aim to define the relationship between the cupric state and oxidative stress in patients, to characterize the involvement of copper in the expression of factors pro-and anti-inflammatory, antioxidant, and mediators of oxidative stress, but also define the role of CFTR on the regulation of inflammatory mediators in the basal state and after a stimulus cupric.
The proposed work will provide new knowledge on the involvement of copper in the process of pulmonary inflammation, and allow to characterize the role of copper transporters in response to oxidative stress. In addition the results will define the role of CFTR on the control of intracellular copper levels. Ultimately, this project could identify new therapeutic targets for chronic inflammatory lung diseases such as cystic fibrosis.
Techniques
Molecular biology - biochemistry - cell biology.
Top page