V. Nivière, C. Caux
The understanding of the catalytic mechanisms of original enzyme systems in relation with their functions within the cells represents a major challenge in modern biology. As part of this problem, my current researches focus on the molecular and structural studies of two different enzyme systems involved in cellular detoxification processes:
• Superoxide reductase SOR, a metalloenzyme involved in the fight against oxidative stress
• MsrPQ, a new discovered system repairing oxidized methionine residues, allowing bacterial resistance to hypochlorous acid (HOCl) stress.
Superoxide reductase SOR: characterization of reaction intermediates Fe3+-OOH and formation of high valent Fe=O species
Some anaerobic and microaerophilic bacteria tolerate the presence of oxygen thanks to a new type of antioxidant enzyme, the superoxide reductase SOR. Discovered in 2000 by our team, SOR catalyzes the detoxification of the superoxide radical O
2•-, a reactive form of oxygen, by a one electron reduction:
O2•- + 1 e- + 2 H+→ H2O2 (SOR)
SOR is a small 14 kDa metalloprotein. Its active site consists of an atypical non-heme mononuclear iron center, coordinated by four nitrogen atoms of histidine in an equatorial plane and a sulfur atom of a cysteine in an axial position (Figure 1):
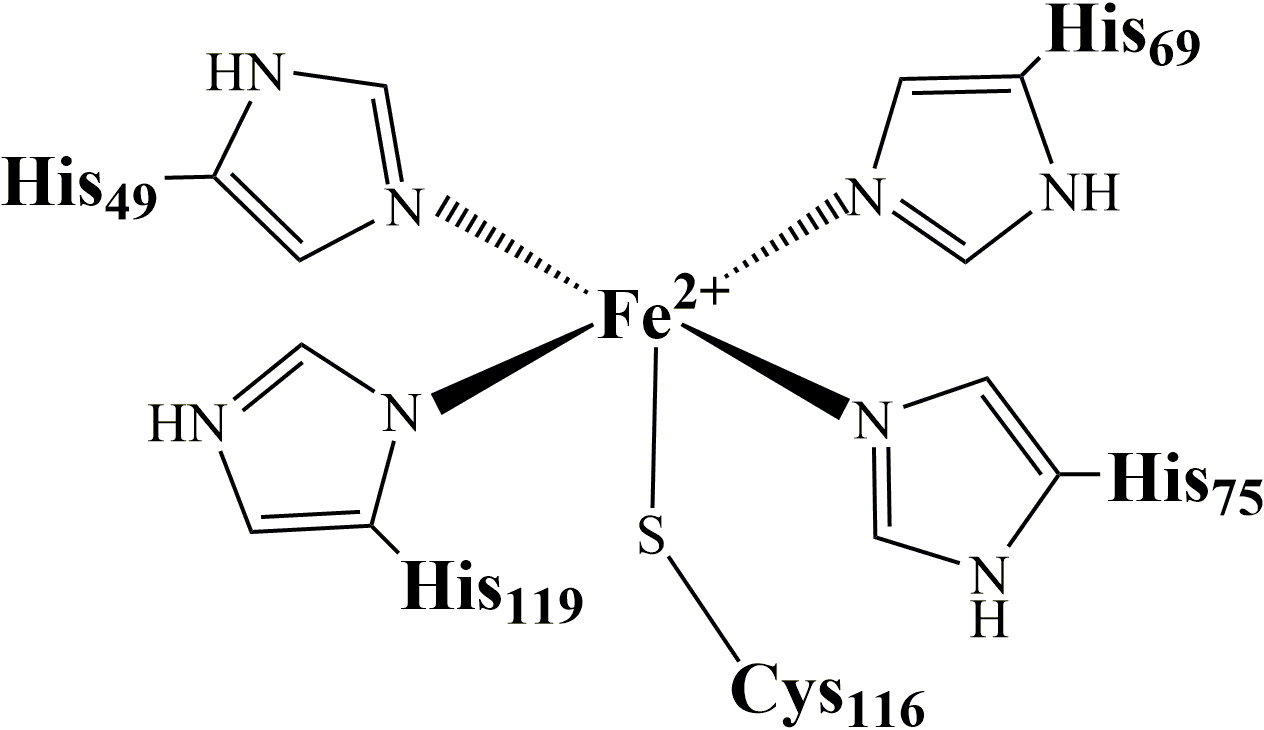
Figure 1: SOR active site.
In the Fe
2+ state, the sixth coordination position is free. It is the site where superoxide binds to be reduced into H
2O
2. This inner sphere reduction mechanism
leads to the formation of a ferric iron hydroperoxide (Fe3+-OOH) intermediate, resulting from the one electron transfer from Fe
2+ to the superoxide, together with a protonation process.
We succeeded in
trapping this intermediary by mutating amino acid residues from the second coordination sphere, which allowed its stabilization. The intermediate was characterized by resonance Raman and
its structure was resolved by X-ray diffraction (Figure 2). This work was published in the journal
Science (
Science, 2007, 316(5823): 449-453):
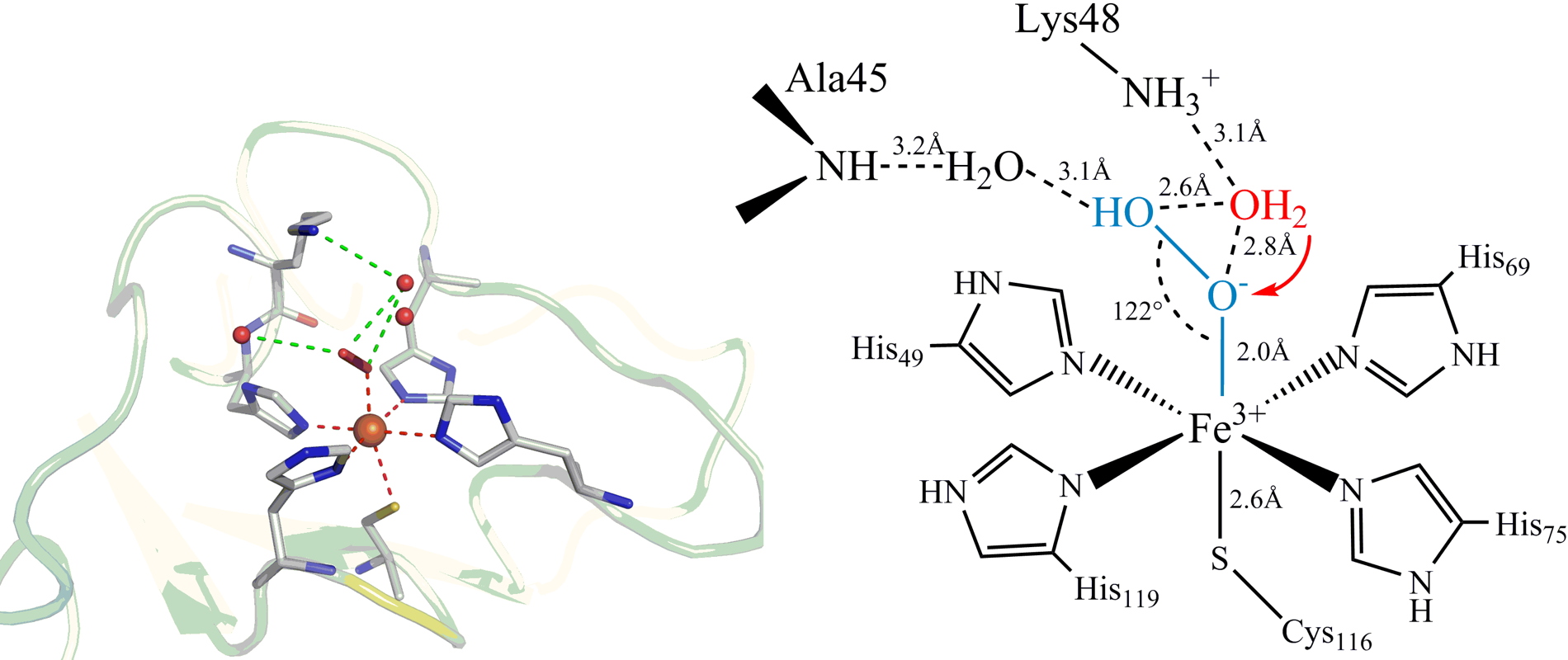
Figure 2: X-ray structure at 1.9 Å resolution of the Fe
3+-OOH intermediate trapped in the SOR active site.
The X-ray structure suggests the involvement of lysine 48 in the protonation of the proximal oxygen of Fe
3+-OOH to form the final product H
2O
2. This second protonation process appears to be one of the key points in the SOR reaction mechanism.
Katona G, Carpentier P, Niviere V, Amara P, Adam V, Ohana J, Tsanov N and Bourgeois D. Raman-assisted crystallography reveals end-on peroxide intermediates in a nonheme iron enzyme.
Science, 2007,
316(5823): 449-453
Control of the evolution of the Fe3+-OOH intermediate and formation of an Iron-oxo species in SOR The SOR active site presents remarkable similarities to that of cytochrome P450-type oxygenases: (i) The coordination of the iron is identical in these two enzymes, FeN
4S
1 type, with a same geometry; (ii) These two enzymes form the same Fe
3+-OOH reaction intermediate during their catalytic cycle.
However, in the case of oxygenases, the Fe
3+-OOH intermediate is cleaved between its two oxygen atoms to form a highly reactive species, a high-valent Iron-oxo (Fe=O) species. This Fe=O is can oxidize very stable chemical bonds within different substrates. On the other hand, in the case of SOR, the cleavage of the Fe
3+-OOH intermediate occurs between the iron atom and the proximal oxygen to form hydrogen peroxide:
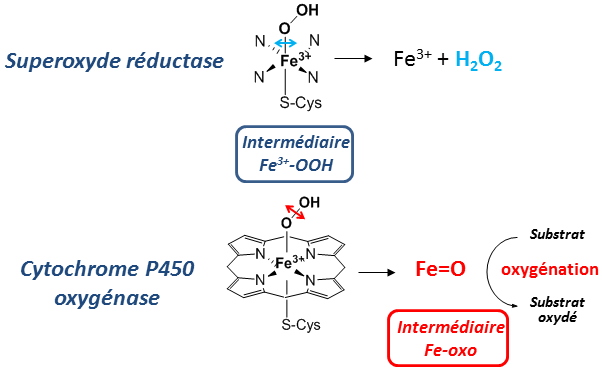
Figure 3: Comparison of the active sites of SOR and cytochrome P450 and difference in the evolution of their Fe
3+-OOH intermediate.
The question is therefore to know
what are the factors which determine the fate of the Fe3+-OOH intermediate in these two systems. Could the presence of the heme in cytochrome P450 or other elements such as residues of the second coordination sphere play a major role?
It is on this issue that we are working on SOR and currently we are studying the role of the protein environment in the evolution of the Fe
3+-OOH intermediate.
Previous works showed that Lysine 48 could be involved in the protonation of the proximal oxygen of the intermediate. Also, Isoleucine 118, whose peptidic NH forms a hydrogen bond with the ligand sulfur of cysteine, could modulate the properties of the Fe
3+-OOH species by trans effect.
By Raman resonance, we have shown that the mutation of each of these two residues leads to the formation of an Iron-oxo species in the SOR active site (Figure 4):
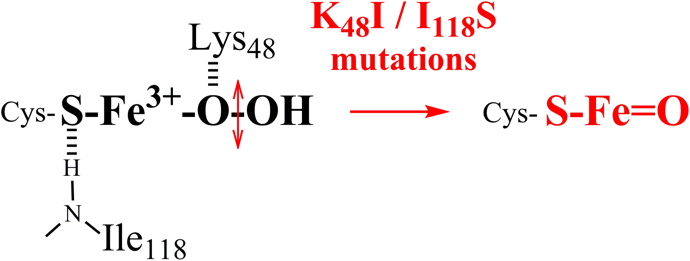
Figure 4: Changes in the iron environment of the SOR active site lead to the formation of an Iron-oxo species.
These results demonstrate the
importance of the second coordination sphere of the SOR ferric site to control the evolution of the Fe3+-OOH intermediate.
This work will be continue along two complementary lines:
• Development of QM-MM theoretical calculations in order to better characterize the elements that lead to the formation of an Iron-oxo species in SOR.
• Studies of other residues of the second coordination sphere for their role in the evolution of the Fe
3+-OOH intermediate.
In addition to the understanding of the catalytic mechanism of SOR, this work also opens up important perspectives for a detailed characterization of the mechanisms of oxygen activation and oxidation catalysis.
 Collaboration
Collaboration Pr. Anne Milet, Groupe Spectrométrie, Interactions, Chimie Théorique, Département de Chimie Moléculaire Université Grenoble Alpes, Grenoble
Publications
David R, Jamet H, Niviere V, Moreau Y and Milet A Iron hydroperoxide intermediate in superoxide reductase: Protonation or dissociation first? MM dynamics and QM/MM metadynamics study.
Journal of Chemical Theory and Computation, 2017,
13(6): 2987-3004
Bonnot F, Tremey E, von Stetten D, Rat S, Duval S, Carpentier P, Clémancey M, Desbois A and Nivière V Formation of high-valent iron-oxo species in superoxide reductase: Characterization by resonance Raman spectroscopy.
Angewandte Chemie International Edition, 2014,
53(23): 5926-5930
Rat S, Ménage S, Thomas F and Nivière V Non-heme iron hydroperoxo species in superoxide reductase as a catalyst for oxidation reactions.
Chemical Communications (Camb), 2014,
50(91): 14213-14216
MsrPQ, a new system to repair oxidized methionines, involved in bacterial resistance to HOCl.
Hypochlorous acid HOCL is a highly oxidizing species, which has been shown to be particularly toxic to microorganisms. It is the active compound in bleach.
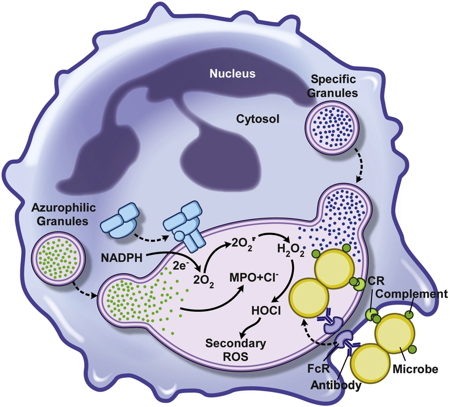
HOCL production is also the first line of defense used by the innate immune system to destroy bacteria and other pathogenic microorganisms. After phagocytosis of pathogens by neutrophils or macrophages, activation of NADPH oxidase NOX leads to a massive intra-phagosomal production of HOCl, which is the main mechanism for eliminating pathogens (Figure 5).
Figure 5: Neutrophil microbicide activity after phagocytosis.
We know that the main targets of HOCl are the methionine residues of bacterial proteins, which are heavily oxidized into methionine sulfoxide (Met-S=O). This type of oxidation causes strong alterations of the protein structures, inducing the loss of their activities as well as their functions. This process leads to the death of the pathogen.
Very recently, in 2015, it appeared that some bacteria express a completely new type of methionine sulfoxide reductase activity (Msr), able of neutralize the effect of HOCl by repairing the oxidized methionines. The singularity of this new Msr activity, called MsrPQ, lies into three main points (Figure 6):
i) Presence of a molybdenum cofactor in the active site of MsrP, while all the Msr characterized so far did not contain metal and functioned according to a chemistry involving a cysteine residue.
ii) Its periplasmic location.
iii) Specific association with a membrane cytochrome, called MsrQ, encoded within the same operon with MsrP.
With this multienzymatic system MsrPQ, bacteria are able to fully reverse the toxic effect of HOCl on periplasmic proteins.
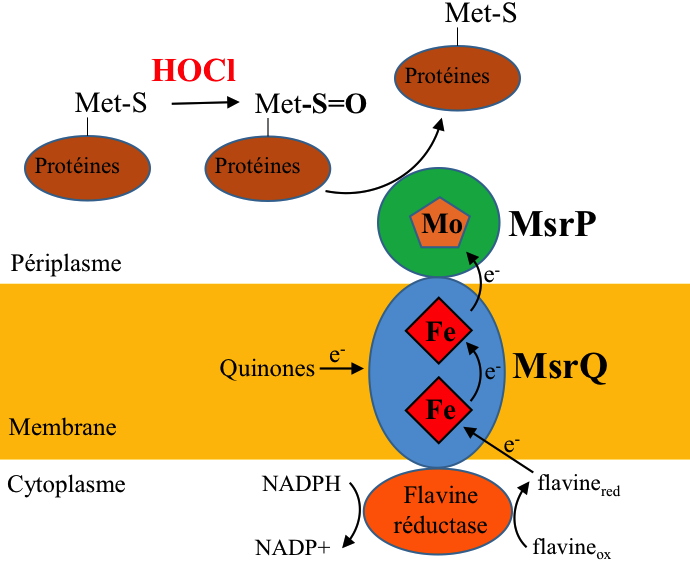
Figure 6: Hypothesis on the organization and functioning of the MsrPQ system to repair oxidized methionines (Met-S=O).
Interestingly, some intracellular pathogenic bacteria present in their genomes a duplication of the MsrPQ operon, which is found in DNA sequences associated with pathogenicity islands. This suggests that the duplication of this operon could have an important role in the virulence of these pathogens.
Our project is to characterize in detail this new MsrPQ system from a molecular and structural point of view and to study the mechanisms of electron transfer, the specific interactions between the different partners, as well as the different aspects that control its overall activity.
Collaboration Pr. Franck Fieschi, Groupe Membrane et Immunité, Institut de Biologie Structurale, 71 avenue des Martyrs, Grenoble
Publication
Juillan-Binard C, Picciocchi A, Andrieu JP, Dupuy J, Petit-Hartlein I, Caux-Thang C, Vives C, Niviere V and Fieschi F A Two-component NADPH Oxidase (NOX)-like system in bacteria is involved in the electron transfer chain to the methionine sulfoxide reductase MsrP.
Journal of Biological Chemistry, 2017,
292(6): 2485-2494