Principal Investigator: Martin J. Field
A long-standing research effort of Martin Field has concerned the development and application of simulation methods for studying biological systems that employ hybrid quantum chemical (QC) or quantum mechanical (QM)/molecular mechanical (MM) approaches. This activity is centred around the open-source molecular modelling software,
Dynamo, which has been under continuous development since the beginning of the 1990s. Dynamo is used by laboratories worldwide, with several of which CoMX has strong collaborations. There remain significant opportunities in making these methods more accurate, computationally efficient and robust, and in adapting them to the study of additional challenging phenomena, such as long-range electron transfer or cases in which an adaptive partitioning between the QC and MM regions is desirable.
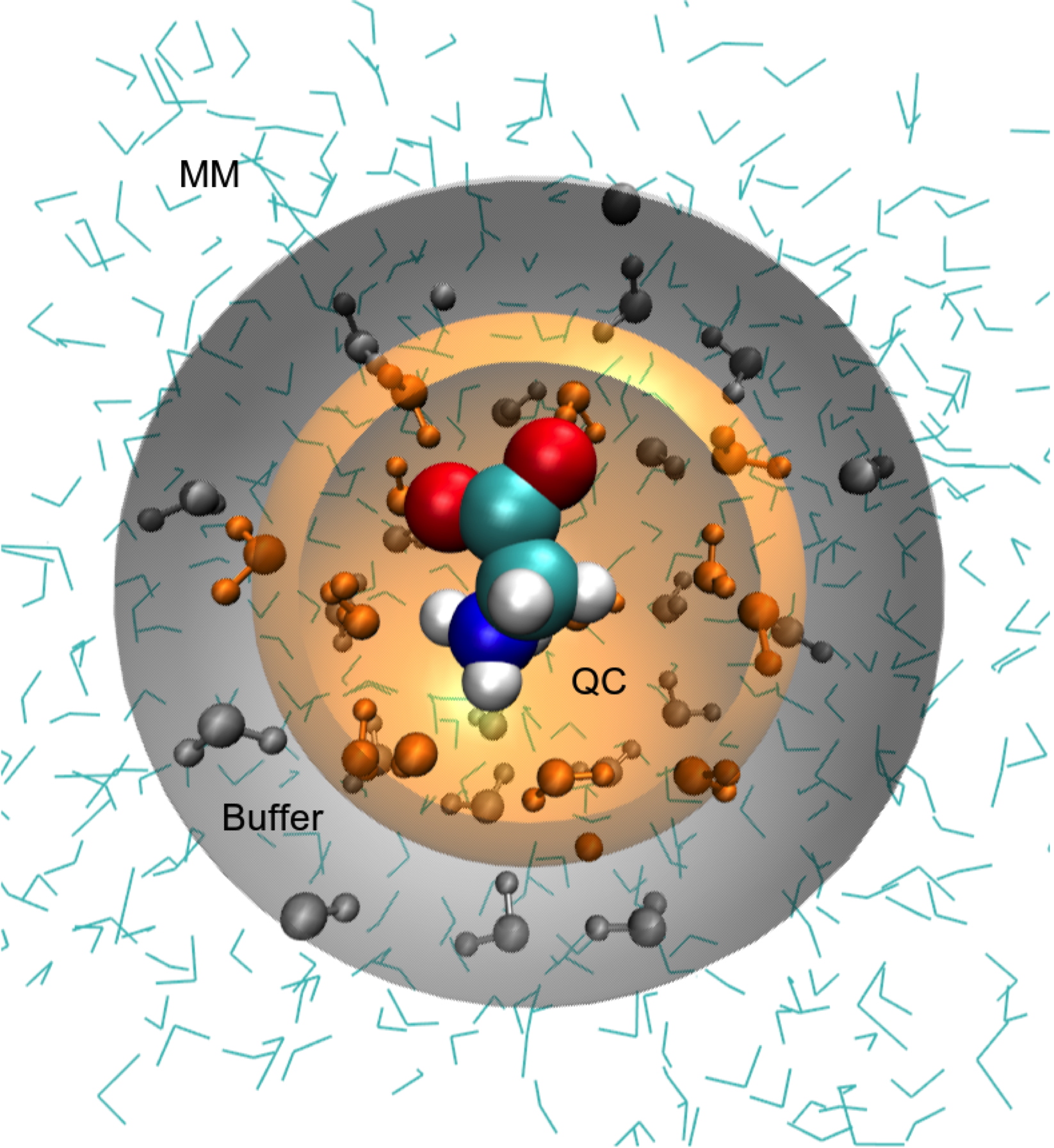 A schematic of the partitioning of a system in an adaptive QC/MM simulation.
A schematic of the partitioning of a system in an adaptive QC/MM simulation.
The solute is treated quantum chemically but a solvent molecule can transform between QC and MM descriptions depending upon its distance from the solute.
From Field MJ. An Algorithm for Adaptive QC/MM Simulations.
Journal of Chemical Theory and Computation, 2017,
13(5): 2342–2351
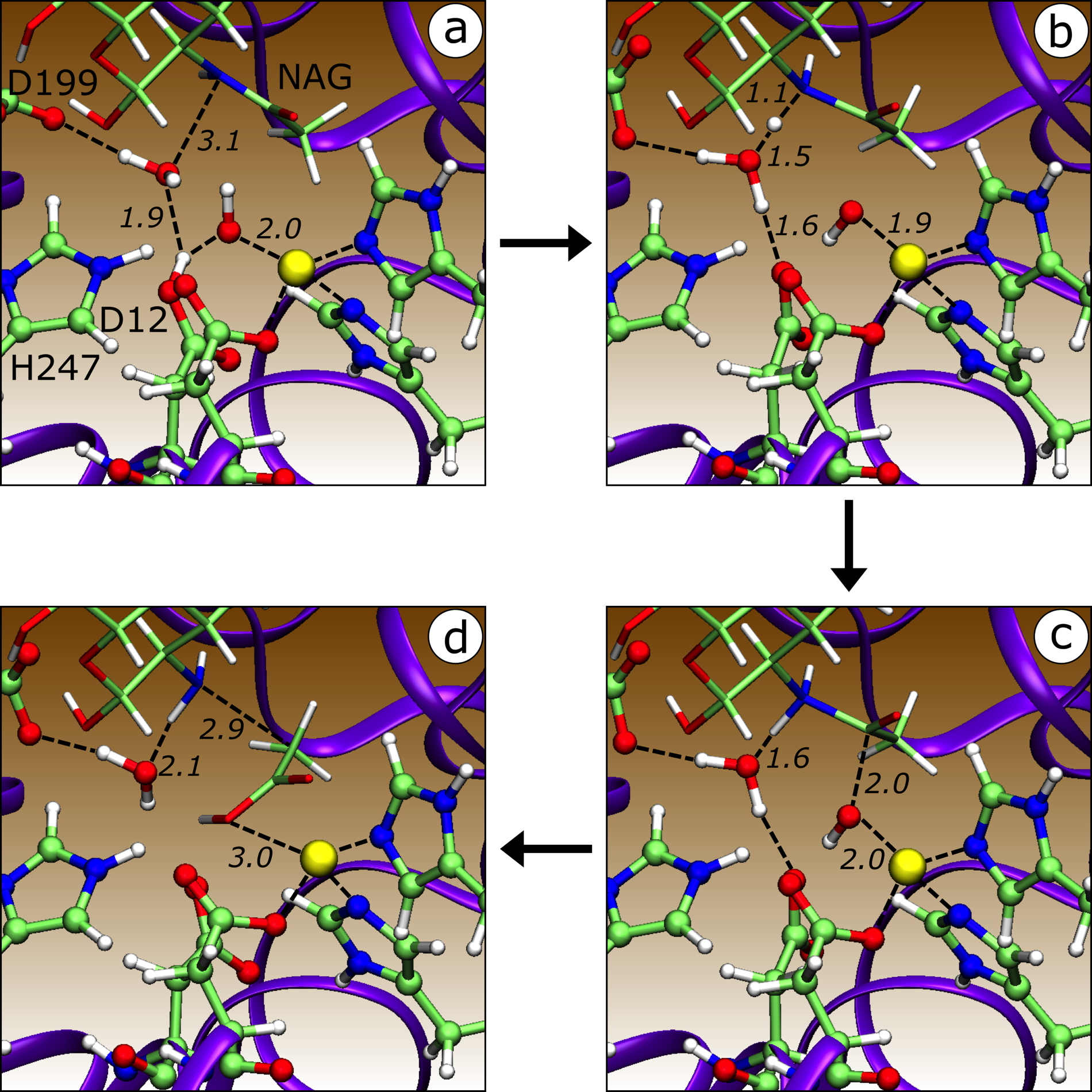 A QC/MM study of the reaction mechanism of the enzyme peptidogylcan deacetylase from Helicobacter pylori.
A QC/MM study of the reaction mechanism of the enzyme peptidogylcan deacetylase from Helicobacter pylori.
The figure shows significant structures along one of the most favorable reaction pathways. These are
(a) the enzyme−substrate complex;
(b) a transition state for a double proton transfer;
(c) the nitrogen-protonated intermediate; and
(d) products (-18 kJ mol
−1 ).
All important residues participating in the reaction are labeled in (a), except for the zinc dication (the yellow sphere), the zinc-bound hydroxide, and the second water in the active site. Unlabeled residues that coordinate the zinc but do not participate in the reaction are Asp14 (in front of Asp12) and His86 and His90 (to the left of the zinc).
From Bhattacharjee N, Feliks M, Shaik MM, Field MJ. Catalytic Mechanism of Peptidoglycan Deacetylase: A Computational Study.
Journal of Physical Chemistry B, 2017,
121(1): 89-99