Dans le cadre restreint d’une étude en mécanique moléculaire (MM), un potentiel empirique définit un champ de force par la donnée d’une fonction d’énergie potentielle Up dans laquelle sont généralement inclus deux types de termes d’interactions
- interactions entre atomes liés (bonded) : liaisons, angles…
- interactions entre atomes non liés (nonbonded) : électrostatiques, Van der Waals.
Les différents termes de l’énergie potentielle Up sont décrits ci après :
Up = Uel + UVdw + Ur + Uθ + UΦ + Uω + UUB
Termes non liés :
- Énergie électrostatique : 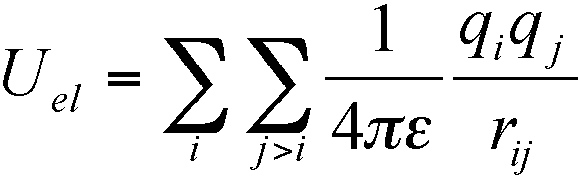 où rij représente la distance entre les atomes i et j et qi, qj, les charges partielles sur les atomes i et j
où rij représente la distance entre les atomes i et j et qi, qj, les charges partielles sur les atomes i et j
- Énergie de Van der Waals : 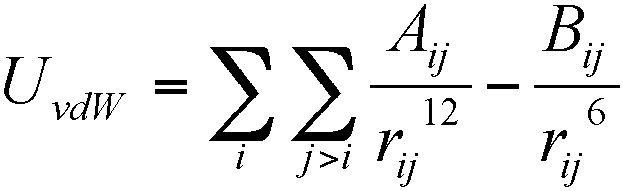 Aij et Bij étant des paramètres définissant profondeur du puits d’attraction entre atomes et distance optimale d’approche entre les atomes i et j.
Aij et Bij étant des paramètres définissant profondeur du puits d’attraction entre atomes et distance optimale d’approche entre les atomes i et j.
 La dynamique moléculaire
La dynamique moléculaire
La Dynamique Moléculaire (DM) est la science de la simulation d'un système de particules. Elle a été appliquée sur des systèmes allant de l'atome jusqu'à une galaxie. La dynamique moléculaire produit des trajectoires c'est à dire les coordonnées de tous les atomes du système en fonction du temps.
L'évolution de ces positions atomiques est décrite par les équations de la mécanique classique de Newton.
En réponse à une force extérieure F, un corps de masse m accélère avec une accélération :
a(t) = F(t) / m
C'est l'équation fondamentale de la dynamique.
F en N = kg m s-2 ; m en kg ; a en m s-2
Dans le cas de forces dites à énergie conservative, la force totale est égale à l'opposé du gradient (les dérivées par rapport aux positions atomiques) de l'énergie potentielle Up précédemment définie
F = - grad Up
Par exemple, pour un terme d’énergie harmonique (ressort s’allongeant de x), Up = 1/2 k x(t)2 et F(t)= m x``(t) = - k x(t), (la force de rappel du ressort).
Cette relation permet de calculer en tout temps t la position de tous les atomes de la molécule étudiée. (Dans le cas du ressort, on tire x(t) de la forme cos (ωt))
La résolution de l’équation de la dynamique nécessite la connaissance de positions et de vitesses initiales. Pour les systèmes biologiques, les coordonnées initiales des atomes proviennent de structures résolues par cristallographie aux rayons X ou RMN et les vitesses initiales sont choisies de manière à représenter la température voulue.
Le résultat d’une simulation de DM consiste en une série de positions atomiques (trajectoire) au cours du temps. La résolution numérique des équations nécessite l’utilisation d’un pas de temps extrêmement petit, de l’ordre de la fs (10-15 s) de manière à prendre en compte les vibrations atomiques les plus rapides (faute de quoi les calculs sont instables). Ceci explique que le temps maximum de simulation dépasse rarement les 10 à 100 ns avec la puissance des ordinateurs actuels, pour des systèmes complexes de plusieurs dizaines de milliers d’atomes (protéines en présence de solvant ou insérées dans des membranes …).
Il faut ensuite exploiter les résultats des calculs en analysant les trajectoires pour aider à la compréhension du mécanisme biologique étudié. On calcule par exemple les fluctuations atomiques, les corrélations entre les mouvements de parties distinctes d’une protéine, le potentiel électrostatique vu par un ligand s’approchant d’une protéine, les modes de vibrations d’une macromolécule…).
Les limitations dans la taille du système (une seule protéine, le plus souvent) et dans le temps de simulation (10 ns) ne sont pas des obstacles incontournables à l’obtention d’information intéressante pour la compréhension de la fonction biologique: le plus souvent, le système est simulé en condition périodique ce qui résout partiellement le problème de la petite taille ; l’étude à l’équilibre de protéines sur des échelles de temps de la nanoseconde, prés de leur structure cristallographique, est souvent très riche en enseignements sur les interactions et corrélations entre résidus. Enfin, il est maintenant courant d’effectuer des calculs d’énergie libre en forçant une transition conformationnelle dans une protéine selon un chemin choisi. Ces calculs sont lourds et limités à l’espace accessible à la protéine sur les échelles de temps de la nanoseconde le long de ce chemin mais ils donnent souvent de très bons résultats directement comparables avec des valeurs expérimentales : δG, barrières d’activation, taux de transition, différences d’affinités…
La modélisation
La modélisation c'est-à-dire le recours à un modèle simplifié pour expliquer un phénomène a trois applications principales en biologie :
Raffinement de structures
- En RMN par la donnée de distances interatomiques (2 à 5 Å) et d’angles dièdres.
- En cristallographie aux rayons X par la donnée de diagrammes de diffraction.
Prédiction de structure
- Prédiction de la structure secondaire ou tertiaire des protéines à partir de leur séquence: Modèles par homologie.
Simulation : une expérimentation numérique in silico complémentaire aux expériences classiques in vitro ou in vivo permettant :
- de calculer des observables expérimentales et de tenter de déduire les mécanismes qui les déterminent
- d'obtenir de grandeurs au-delà des possibilités expérimentales dans des conditions parfaitement contrôlées de pureté, pression, température…
- de tester notre compréhension d'un phénomène à l'échelle atomique
- de visualiser les conséquences de l'agitation thermique sur les structures macromoléculaires (accès de substrats à des sites enzymatiques, transitions conformationnelles, influence de l'eau, couplages entre mouvements.)
Notre activité appartient essentiellement au troisième aspect, c'est-à-dire à l’aspect simulations de macromolécules afin de comprendre certains aspects de leur fonction à partir de leur structure.
Toute molécule étant l'association de divers atomes, la cohésion de cet ensemble résultant des interactions répulsives et attractives entre les atomes va être modélisée par un système purement mécanique de forces, sans prendre en compte la nature microscopique et électrostatique (quantique) des interactions entre les électrons et les noyaux.
Une macromolécule est ainsi modélisée par un réseau de masses ponctuelles chargées en leur centre, reliées par des ressorts et interagissant via des potentiels empiriques.