RNA biosynthesis involves many biochemical events that are essential for its maturation. From an enzymatic point of view, the nucleoside modification phenomenon is certainly one of the most remarkable of these events. It allows a multitude of structural changes in the canonical bases adenosine, guanosine, cytosine and uridine. A number of these modifications, which result from complex processes (hyper-modifications), have been characterized and are mainly located at positions 34 and 37 of transfer RNAs (tRNAs) in the so-called "anticodon" loop (
Figure). They are particularly interesting because they are known to optimize the decoding function of messenger RNA by directly controlling codon-anticodon interactions. Enzymes catalyzing nucleoside modifications are fundamental targets for research, as they allow the study of protein-RNA interactions and are implicated in some diseases. Despite the wealth of genetic, physiological and biochemical data on the tRNA-modifying enzymes, very little structural data are available in the literature.
For several years, we have been studying the enzymes involved in the biosynthetic pathway leading to the formation of the hyper-modified nucleoside ms
2io
6A37. This hyper-modification results from a sequence of enzymatic reactions successively catalyzed by the enzymes
MiaA,
MiaB and
MiaE, the last two of which belong to the class of metallo-enzymes. By combining biochemical, spectroscopic and crystallographic methods, we have identified the structural elements of MiaE that allow the docking of the enzyme with tRNA and the channeling of molecular oxygen, both of which are essential primo-processes that initiate the hydroxylation transformation.
We obtained the crystallographic structure of the
Pseudomonas putida MiaE enzyme (pp-MiaE). This structure is in the form of a single, compact domain that consists essentially of a bundle of four antiparallel α-helices housing a catalytic center with two iron atoms. These two metal ions are coordinated by histidines and glutamates, and bridged by an oxygen atom characterized as "μ-oxo" by spectroscopy. Two functional channels were identified. The first was mapped using krypton atoms (oxygen substitute) introduced under pressure into Pp-MiaE crystals. This channel of hydrophobic in nature, is undoubtedly the diffusion route of molecular oxygen to the active site for catalysis. The second channel, polar by nature, is interpreted as a large cavity to stereoselectively accommodate the anti-codon part of the tRNA and to position the adenine base 37 close to the active site to facilitate the hydroxylation of its isopentenyl moiety. A structural model of the MiaE-tRNA complex was simulated based on the crystallographic structures, the biochemical and spectroscopic results. This model reveals that a non-canonical flexible loop of the MiaE enzyme, is likely involved in tRNA recognition and docking processes.
Together, these results pave the way for understanding the mechanisms involved in enzyme-substrate recognition in tRNA modification reactions.
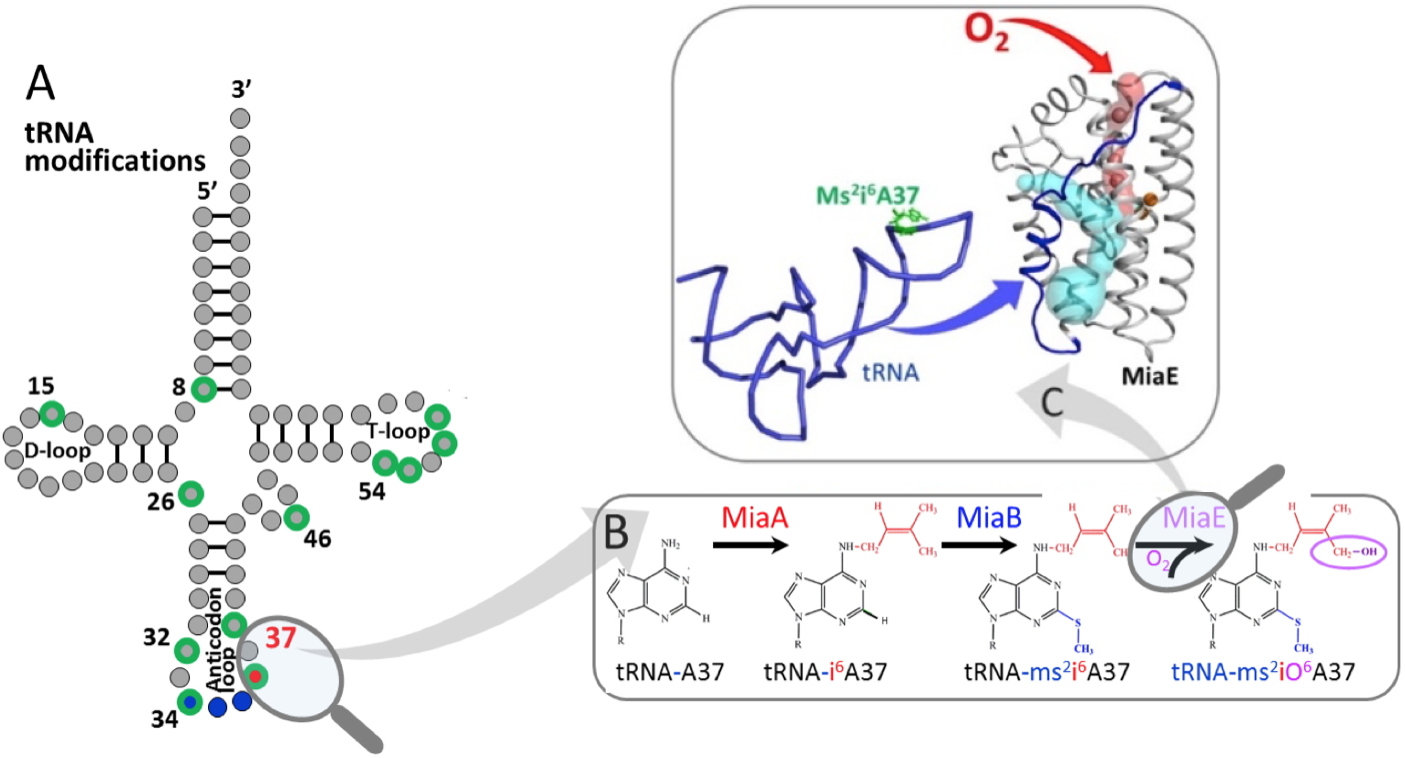 A - Typical modifications of tRNAs (in green), including those at position 37 on the anticodon loop.
A - Typical modifications of tRNAs (in green), including those at position 37 on the anticodon loop.
B - Details of the A37 hyper-modifications of tRNA catalyzed successively by the MiaA, MiaB and MiaE enzymes.
C - Structure of the MiaE enzyme, the oxygen channel is in red, the cavity housing the tRNA is in light blue. The tRNA is in blue and the modified base is in green.
This hyper-modified nucleoside is 2-methylthio-N6-(cis-hydroxy)isopentenyl-adenosine (ms2io6A37) which is present at position 37 adjacent to the anticodon of some tRNAs.
MiaA allows the addition of the isopentenyl group on the N6 exocyclic nitrogen of adenosine, converting tRNA-A37 to tRNA-i6A37.
MiaB is an enzyme of the Radical-SAM family that catalyzes the insertion of the thiomethyl group at position 2, converting tRNA-i6A37 to tRNA-ms2i6A37.
MiaE belongs to the family of proteins with a non-heme iron center. This family consists of extremely diverse enzymes, identified in all kingdoms of life, which catalyze an impressive number of different chemical oxidations.