Theoretical chemistry, a branch of physical chemistry using quantum mechanics equations implemented in computer programs, is used to determine the structures and properties of chemical systems at the atomic and molecular level. This approach makes possible the determination of energetic, geometrical and electronic properties of systems studied. Among the results provided, one is able to determine the reaction path followed along a chemical process (mechanisms, reaction energies and activation barriers) or simulate spectroscopic data (UV/Vis, infrared) of a given molecule. Theoretical chemistry is thus placed both upstream (prediction and prospection) and downstream (explanation) of the experiment, to which it comes as a natural complement. A new simulation protocol using theoretical chemistry methods has been developed and applied by researchers at our laboratory. This approach aims to accurately simulate the reactivity of metalloenzymes by meta-dynamics
QM/MM* methods. The goal is to build the free energy profile of a reaction process taking place within a metalloenzyme, while taking into account the dynamic effects of the environment such as the enzymatic structure and the solvent (
Figure).
The developed methodology was used to elucidate the role of the environment during the second protonation step that occurs during the catalytic cycle of the superoxide reductase (SOR) enzyme, whose activity is to detoxify the cells by reducing superoxide into in hydrogen peroxide
2. Experimentally, it has been shown that this step was decisive for the enzymatic activity. Understanding the native mechanism is therefore fundamental to explain the role of the active site environment on the activity of the enzyme and also to consider modifying this activity. The developed protocol involves simulations of classical molecular dynamics to study the structuration and micro-solvation of the active site, as well as the interaction of neighboring residues with the substrates and their conformational properties. QM/MM
meta-dynamics simulations were then used to explore the different possible reactive pathways for H
2O
2 formation. Among the results obtained, the influence of the neighboring residues, together with the solvent effect, have been shown to be of particular relevance since the second protonation is preferably carried out via an unexpected mechanism.
This innovative approach in several aspects (complete protocol, high level of calculation for the quantum part, simulation time) made it possible to clarify certain fundamental aspects of the reactivity of the SOR. It is now applied to the study of different mutants, to explain how certain mutations affect the reactivity of the enzyme.
QM/MM (Quantum Mechanics / Molecular Mechanics) is a multi-scale approach in which the active site of an enzyme is described by quantum mechanics to simulate chemical reactivit (bond formation/breakage) while the environment is described by molecular mechanics. The development of QM/MM methods was awarded in 2013 by the Nobel Prize in Chemistry.
Meta-dynamics is a method that simulates the dynamic evolution of a chemical system while exploring in a privileged way, a specific coordinate related to the reaction studied. It thus makes possible to determine a free energy profile for the reaction path leading from the reagent to the product and decipher with a high level of precision the chemical mechanisms at work.
These results were obtained by Rolf David during his PhD (LabEx ARCANE funding), in a collaboration between the Chemistry and Biology of Metals laboratory, and the Department of Molecular Chemistry of Grenoble-Alpes University.
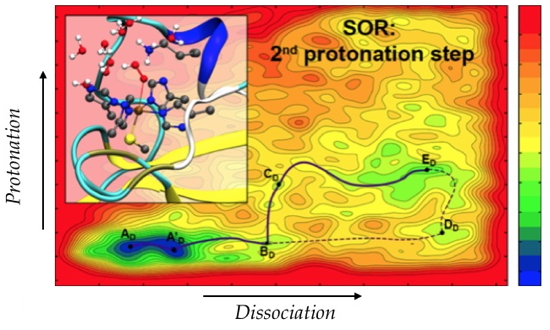
Reaction path determined by meta-dynamics QM/MM.